Monoclonal Antibodies in Preclinical EAE Models of Multiple Sclerosis: A Systematic Review


Abstract
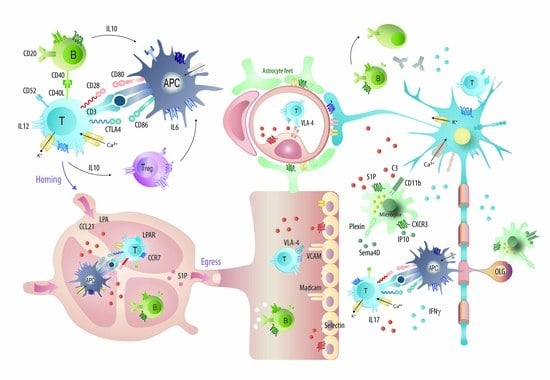
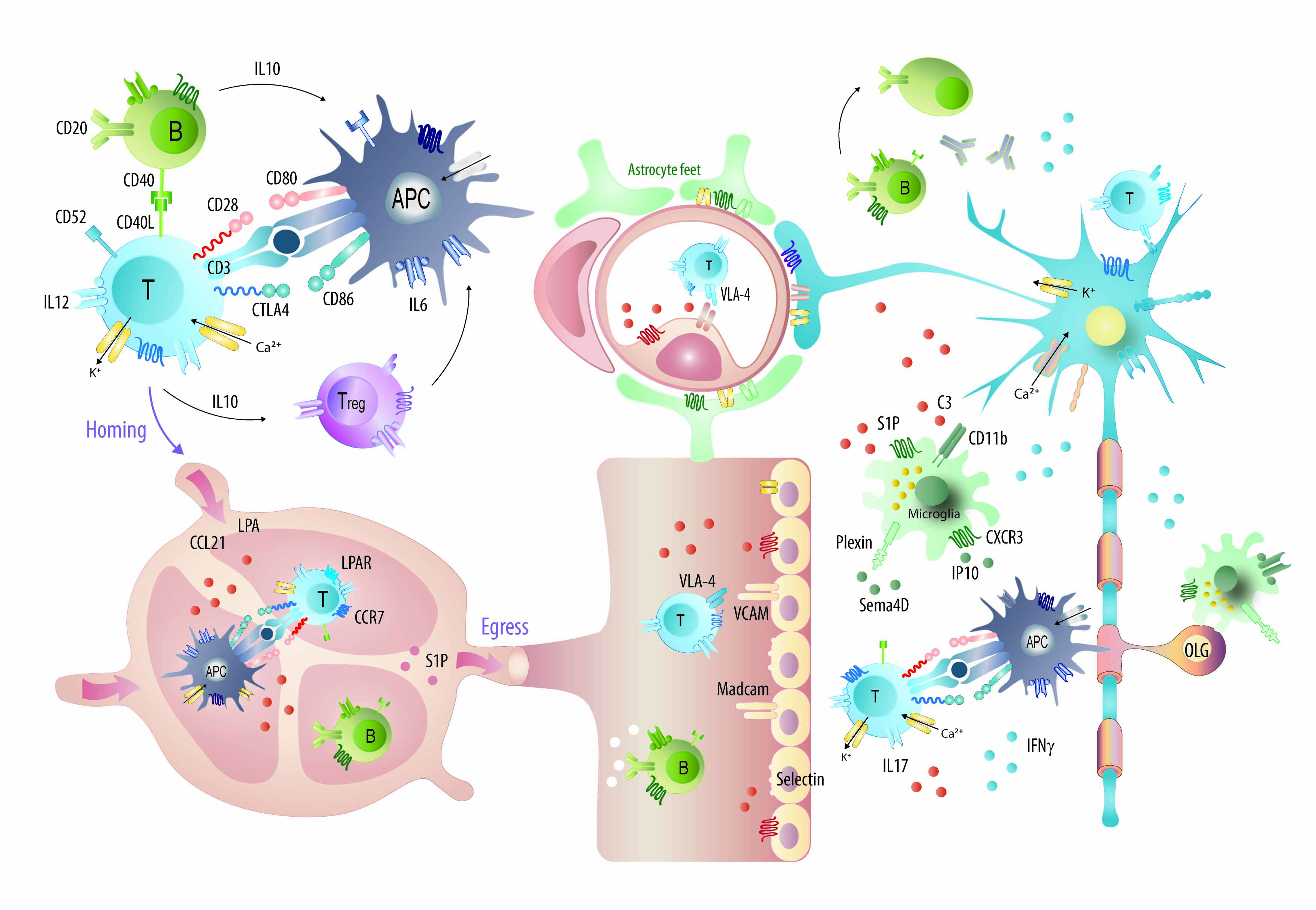
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
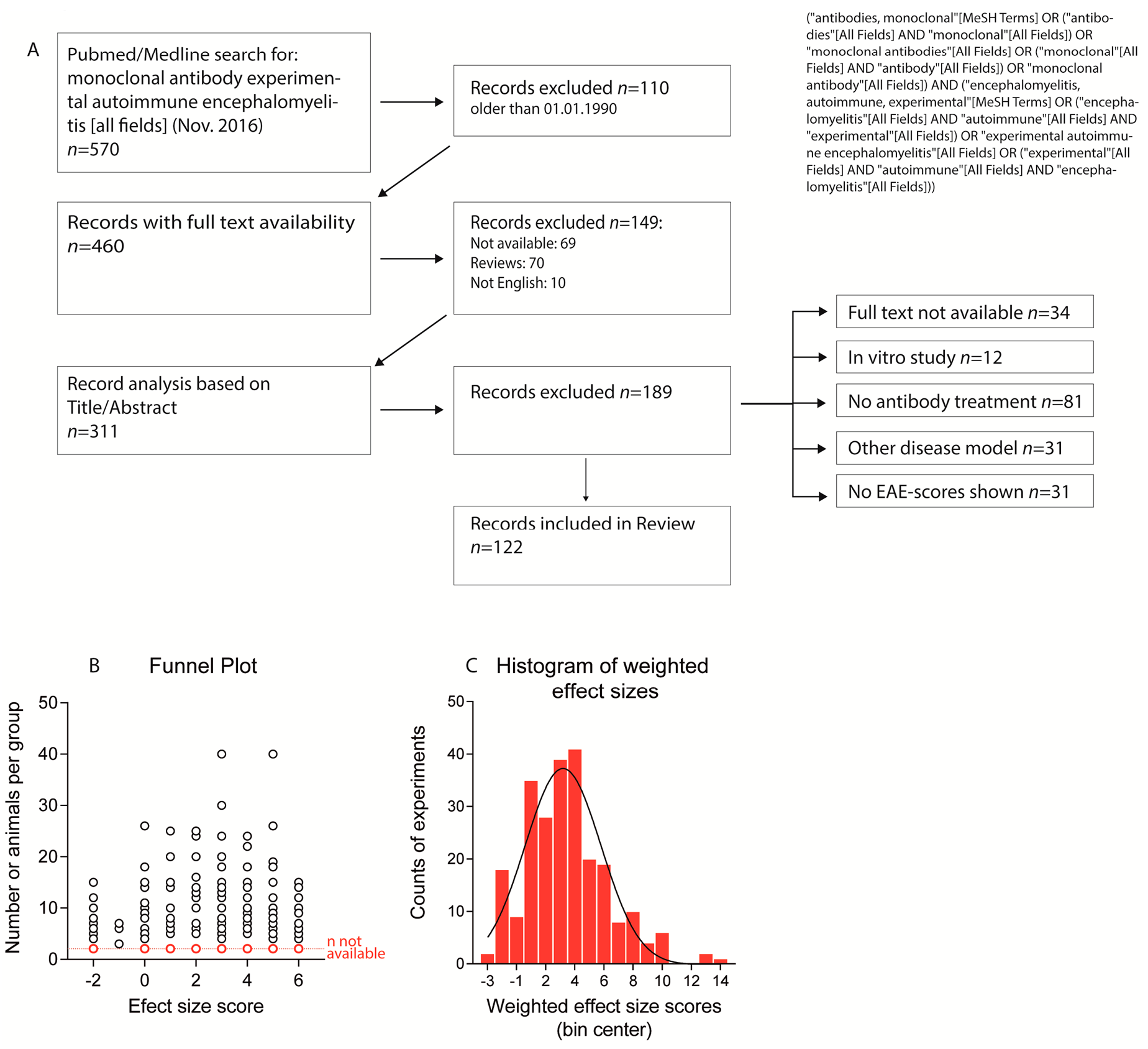
2.1. Paper Evaluation
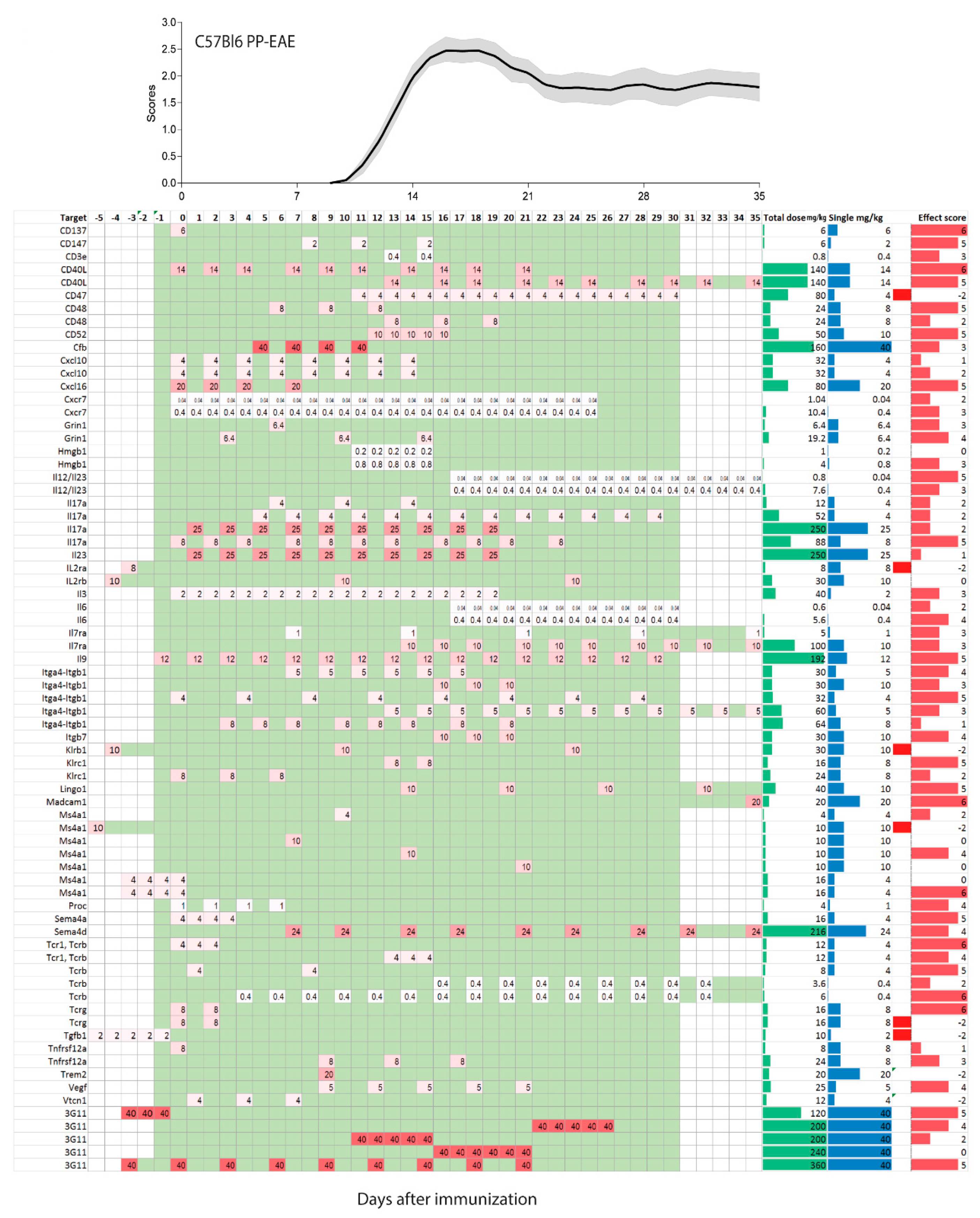
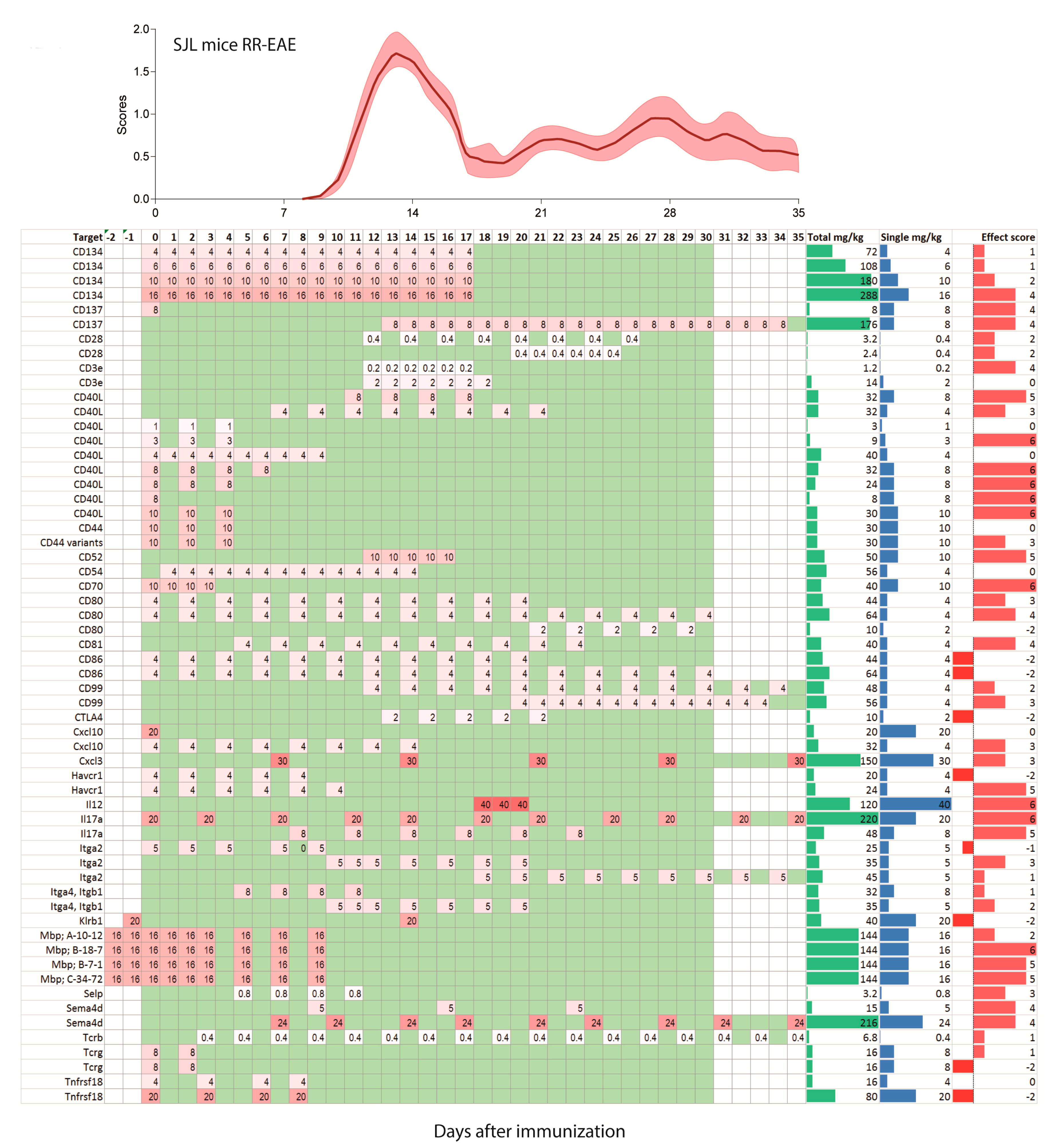
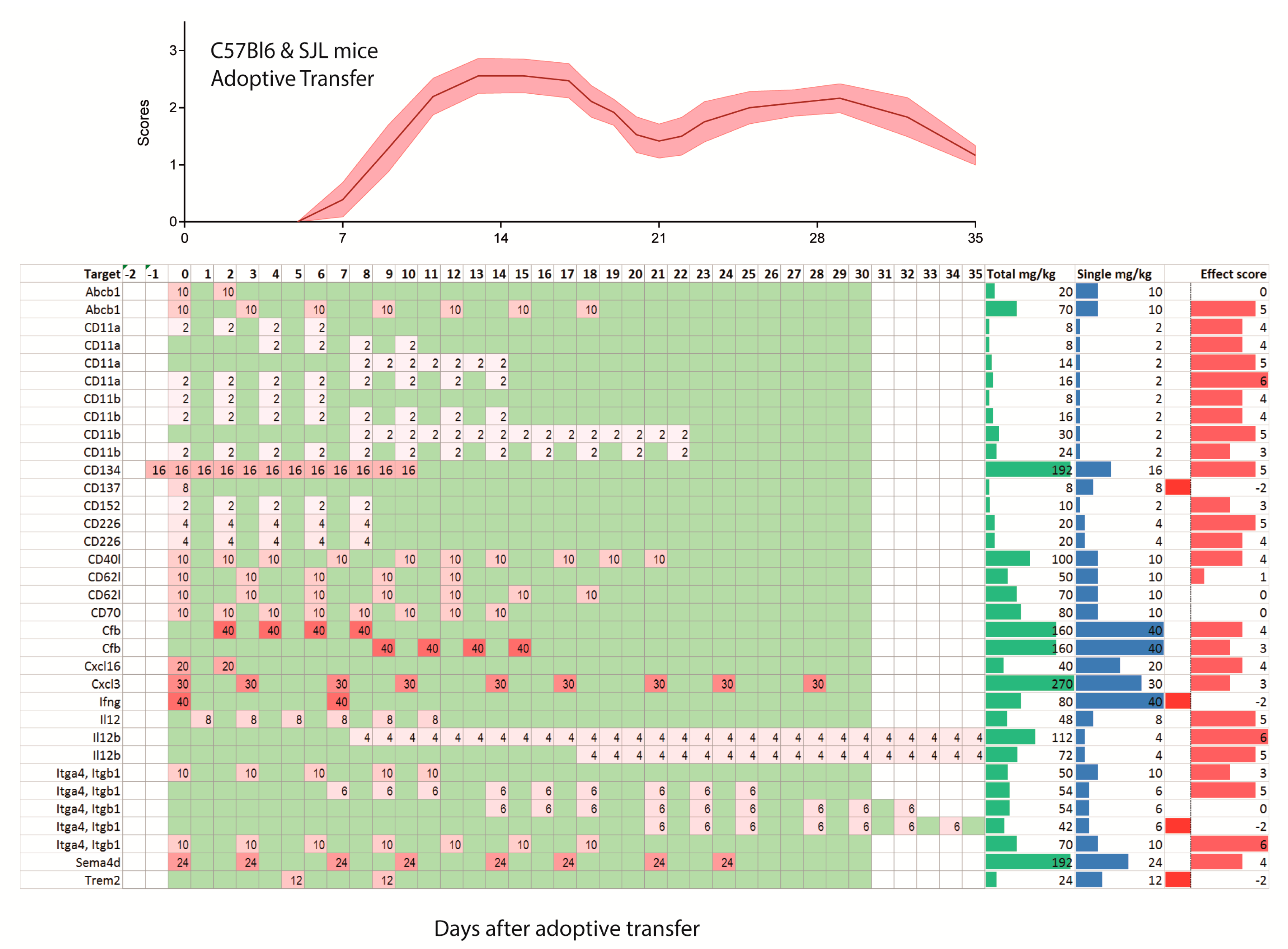
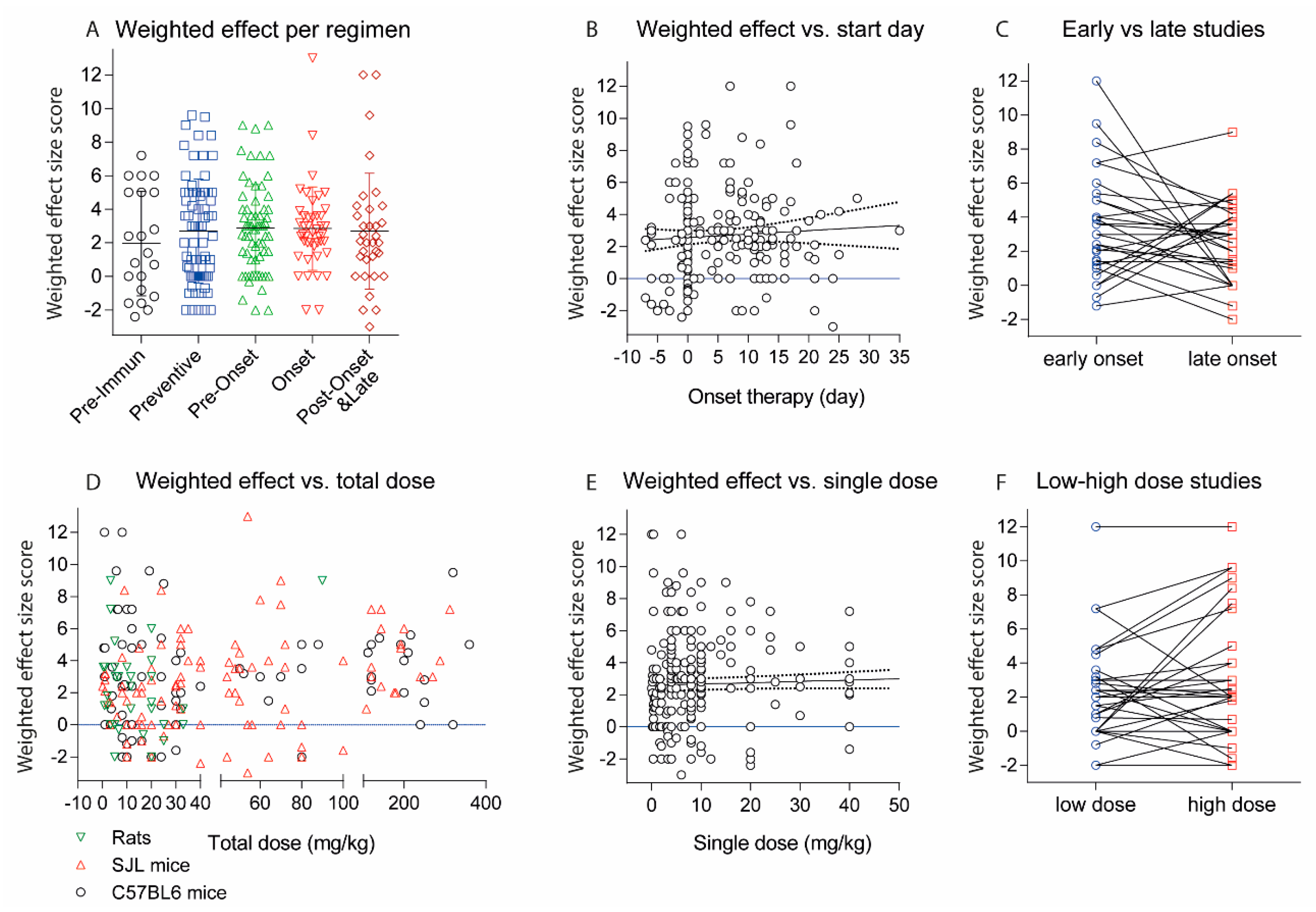
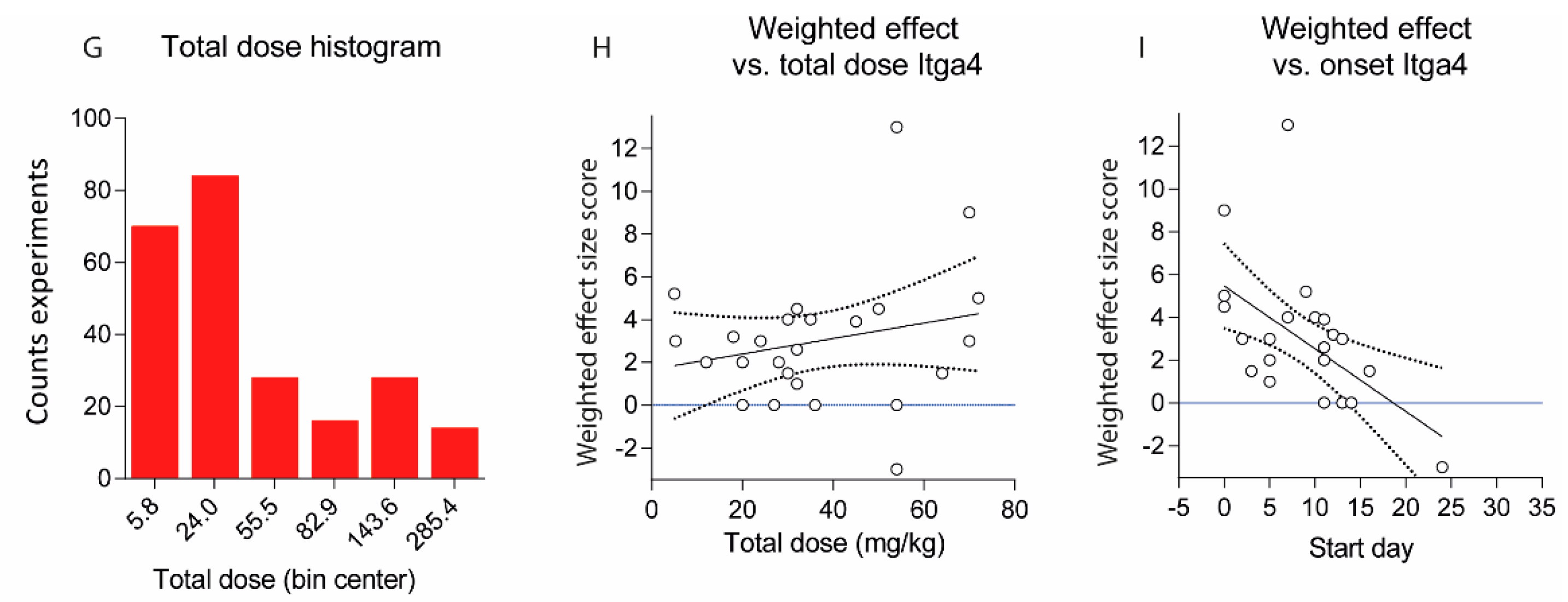
2.2. Treatments Schedules and Doses
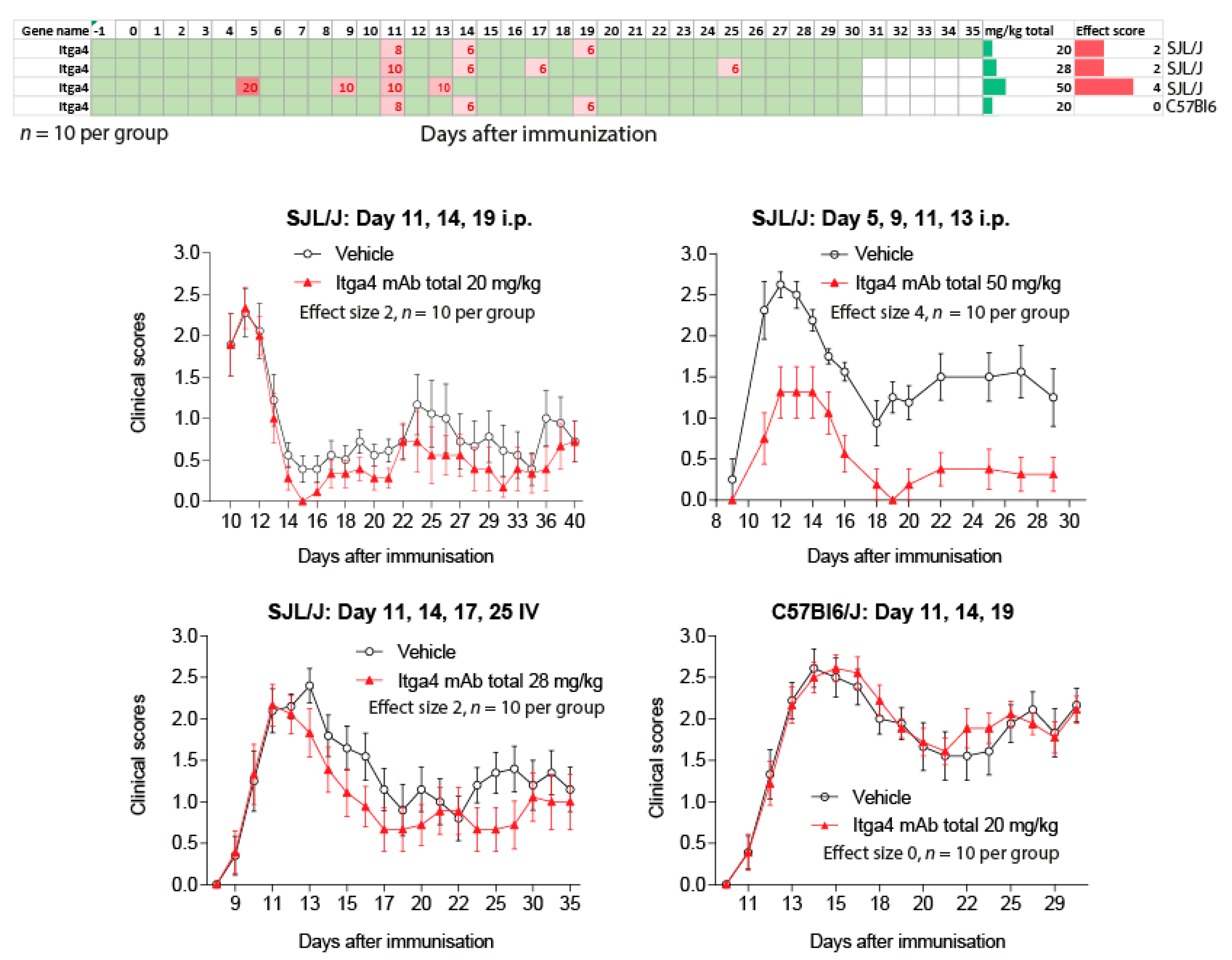
2.3. Natalizumab Effects Depending on Treatment Schedules
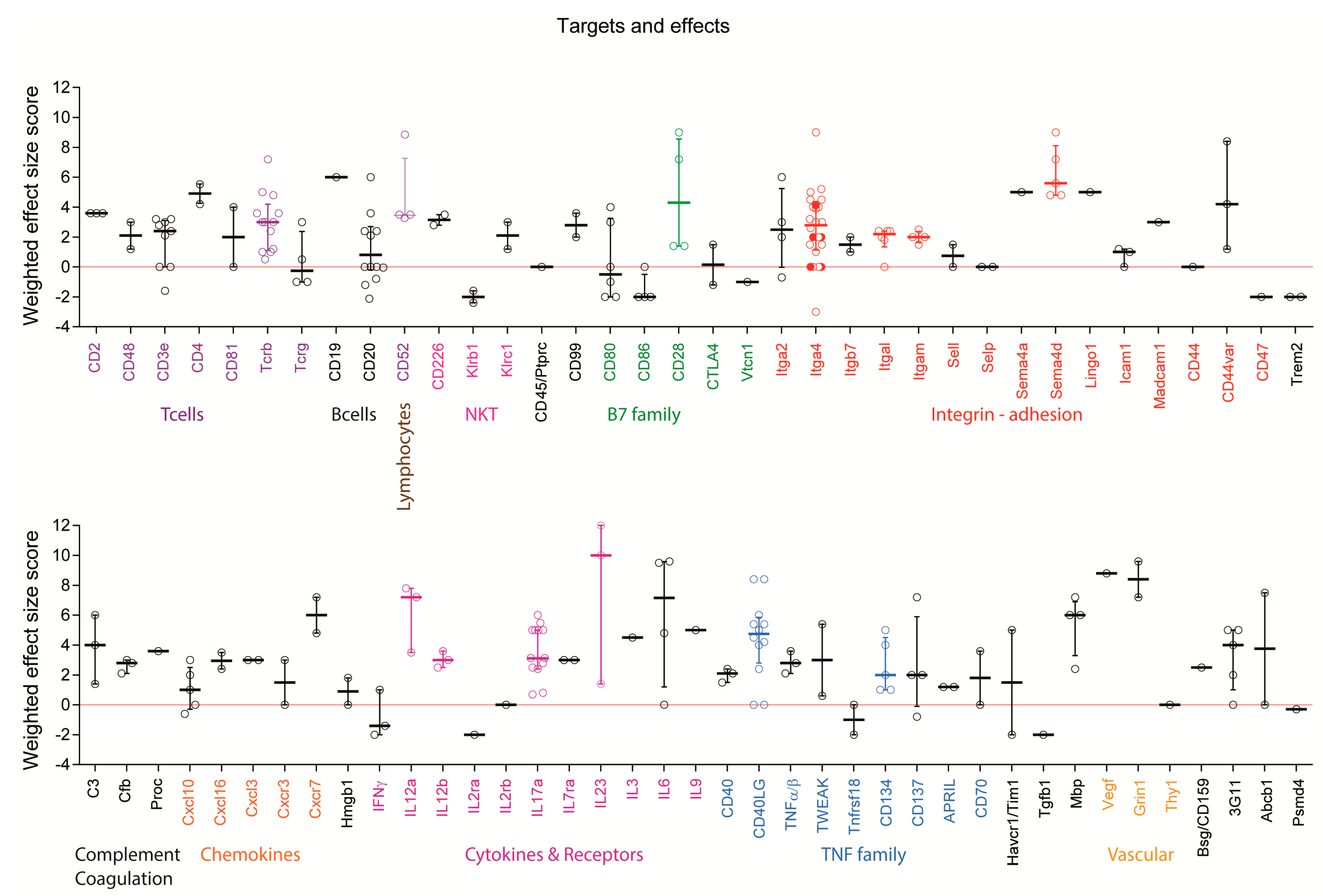
2.4. Targets: Favorable Candidates
2.5. Targets: Unfavorable Candidates
2.6. Experimental Autoimmune Encephalomyelitis (EAE) Models and Strains or Species Effects
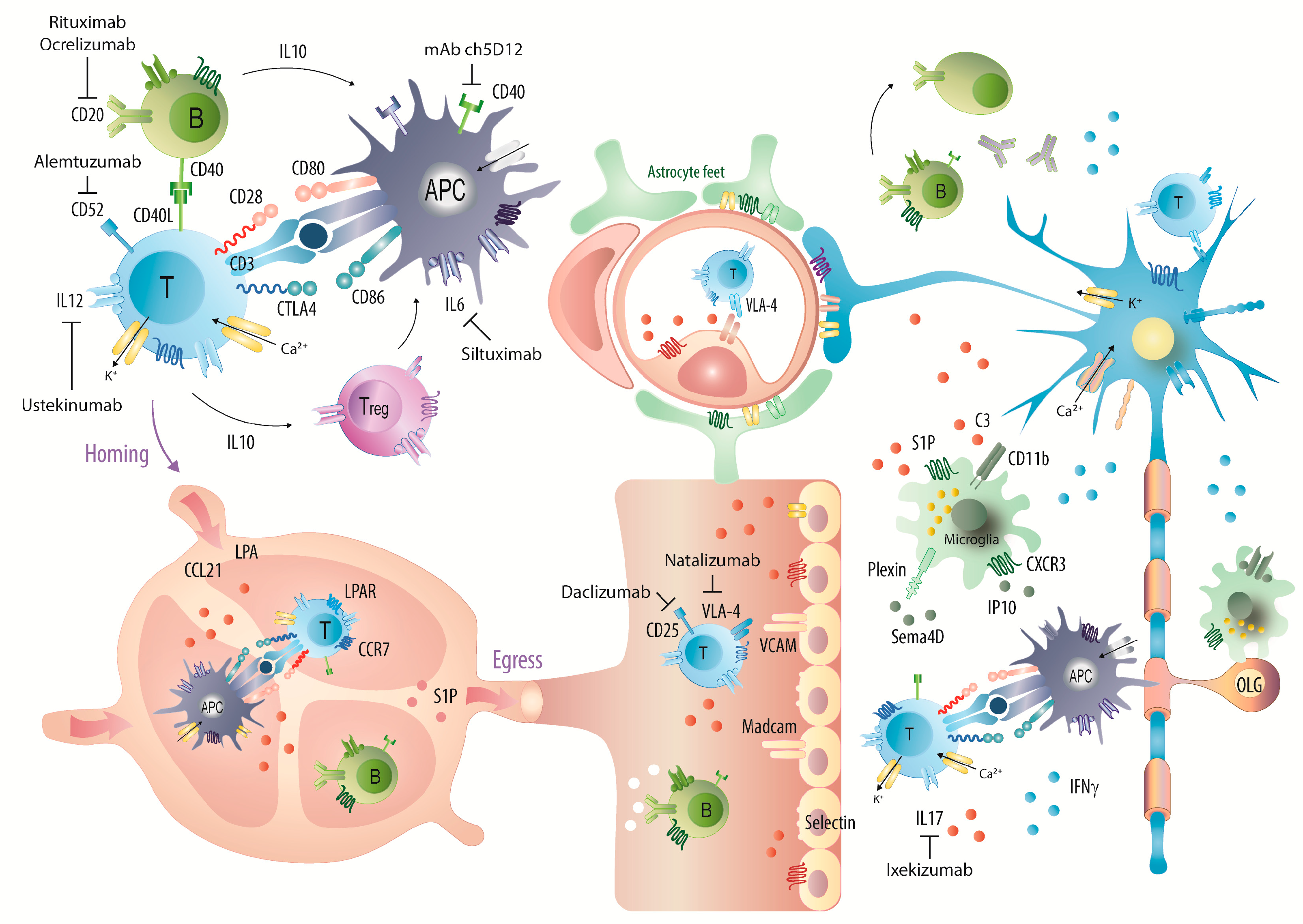
3. Discussion
4. Methods
4.1. Literature Search, Dosing Estimates and Association Analysis
4.2. Induction of EAE and Clinical Assessment of the Schedule Dependent Effects of Anti-Itga4
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab vs. Interferon β-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab vs. Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Rommer, P.S.; Dudesek, A.; Stuve, O.; Zettl, U.K. Monoclonal antibodies in treatment of multiple sclerosis. Clin. Exp. Immunol. 2014, 175, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Dahlhaus, S.; Hoepner, R.; Chan, A.; Kleiter, I.; Adams, O.; Lukas, C.; Hellwig, K.; Gold, R. Disease course and outcome of 15 monocentrically treated natalizumab-associated progressive multifocal leukoencephalopathy patients. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.D.; Robertson, N.P. Alemtuzumab for multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2016, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A.; Coles, A.J.; Sullivan, H.; Fox, E.; Goldberg, M.; Oyuela, P.; Purvis, A.; Beardsley, D.S.; Margolin, D.H. A distinctive form of immune thrombocytopenia in a phase 2 study of alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Blood 2011, 118, 6299–6305. [Google Scholar] [CrossRef] [PubMed]
- Howard, L.M.; Miga, A.J.; Vanderlugt, C.L.; dal Canto, M.C.; Laman, J.D.; Noelle, R.J.; Miller, S.D. Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J. Clin. Investig. 1999, 103, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Durell, B.G.; Miga, A.V.; Hickey, W.F.; Noelle, R.J. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J. Exp. Med. 2001, 193, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerken, L.; Haspels, I.; van Rijs, W.; Blauw, B.; Ferrant, J.L.; Hess, D.M.; Garber, E.A.; Taylor, F.R.; Burkly, L.C. FcR interactions do not play a major role in inhibition of experimental autoimmune encephalomyelitis by anti-CD154 monoclonal antibodies. J. Immunol. 2004, 173, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Jagessar, S.A.; Heijmans, N.; Oh, L.; Bauer, J.; Blezer, E.L.; Laman, J.D.; Migone, T.S.; Devalaraja, M.N.; A’t Hart, B. Antibodies against human BLyS and APRIL attenuate EAE development in marmoset monkeys. J. Neuroimmune Pharmacol. 2012, 7, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laman, J.D.; Maassen, C.B.; Schellekens, M.M.; Visser, L.; Kap, M.; de Jong, E.; van Puijenbroek, M.; van Stipdonk, M.J.; van Meurs, M.; Schwarzler, C.; et al. Therapy with antibodies against CD40L (CD154) and CD44-variant isoforms reduces experimental autoimmune encephalomyelitis induced by a proteolipid protein peptide. Mult. Scler. 1998, 4, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Roy, A.; Pahan, K. Functional blocking monoclonal antibodies against IL-12p40 homodimer inhibit adoptive transfer of experimental allergic encephalomyelitis. J. Immunol. 2009, 182, 5013–5023. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, H.H.; Ibrahim, S.M.; Koczan, D.; Kruse, N.; Weishaupt, A.; Toyka, K.V.; Gold, R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell. Immunol. 2005, 237, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Mitsdoerffer, M.; Xiao, S.; Gu, G.; Sobel, R.A.; Kuchroo, V.K. IL-21R signaling is critical for induction of spontaneous experimental autoimmune encephalomyelitis. J. Clin. Investig. 2015, 125, 4011–4020. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2016, 2, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Mix, E.; Meyer-Rienecker, H.; Zettl, U.K. Animal models of multiple sclerosis for the development and validation of novel therapies—Potential and limitations. J. Neurol. 2008, 255, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Mix, E.; Meyer-Rienecker, H.; Hartung, H.P.; Zettl, U.K. Animal models of multiple sclerosis—Potentials and limitations. Prog. Neurobiol. 2010, 92, 386–404. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L.; Zamvil, S.S. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005, 26, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Monson, N.L.; Cravens, P.; Hussain, R.; Harp, C.T.; Cummings, M.; de Pilar Martin, M.; Ben, L.H.; Do, J.; Lyons, J.A.; Lovette-Racke, A.; et al. Rituximab therapy reduces organ-specific T cell responses and ameliorates experimental autoimmune encephalomyelitis. PLoS ONE 2011, 6, e17103. [Google Scholar] [CrossRef] [PubMed]
- Robert, R.; Ang, C.; Sun, G.; Juglair, L.; Lim, E.X.; Mason, L.J.; Payne, N.L.; Bernard, C.C.; Mackay, C.R. Essential role for CCR6 in certain inflammatory diseases demonstrated using specific antagonist and knockin mice. JCI Insight 2017, 2, e94821. [Google Scholar] [CrossRef] [PubMed]
- Moreno Torres, I.D.; Garcia-Merino, A. Anti-CD20 monoclonal antibodies in multiple sclerosis. Expert Rev. Neurother. 2016, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bosl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Laman, J.D.; A’t Hart, B.; Brok, H.; Meurs, M.; Schellekens, M.M.; Kasran, A.; Boon, L.; Bauer, J.; Boer, M.; Ceuppens, J. Protection of marmoset monkeys against EAE by treatment with a murine antibody blocking CD40 (mu5D12). Eur. J. Immunol. 2002, 32, 2218–2228. [Google Scholar] [CrossRef]
- Boon, L.; Brok, H.P.; Bauer, J.; Ortiz-Buijsse, A.; Schellekens, M.M.; Ramdien-Murli, S.; Blezer, E.; van Meurs, M.; Ceuppens, J.; de Boer, M.; et al. Prevention of experimental autoimmune encephalomyelitis in the common marmoset (Callithrix jacchus) using a chimeric antagonist monoclonal antibody against human CD40 is associated with altered B cell responses. J. Immunol. 2001, 167, 2942–2949. [Google Scholar] [CrossRef] [PubMed]
- Brok, H.P.; van Meurs, M.; Blezer, E.; Schantz, A.; Peritt, D.; Treacy, G.; Laman, J.D.; Bauer, J.; A’t Hart, B. Prevention of experimental autoimmune encephalomyelitis in common marmosets using an anti-IL-12p40 monoclonal antibody. J. Immunol. 2002, 169, 6554–6563. [Google Scholar] [CrossRef] [PubMed]
- Mullin, A.P.; Cui, C.; Wang, Y.; Wang, J.; Troy, E.; Caggiano, A.O.; Parry, T.J.; Colburn, R.W.; Pavlopoulos, E. rHIgM22 enhances remyelination in the brain of the cuprizone mouse model of demyelination. Neurobiol. Dis. 2017, 105, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Kap, Y.S.; Jagessar, S.A.; van Driel, N.; Blezer, E.; Bauer, J.; van Meurs, M.; Smith, P.; Laman, J.D.; A’t Hart, B. Effects of early IL-17A neutralization on disease induction in a primate model of experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 2011, 6, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuno, T.; Nakatsuji, Y.; Moriya, M.; Takamatsu, H.; Nojima, S.; Takegahara, N.; Toyofuku, T.; Nakagawa, Y.; Kang, S.; Friedel, R.H.; et al. Roles of Sema4D-plexin-B1 interactions in the central nervous system for pathogenesis of experimental autoimmune encephalomyelitis. J. Immunol. 2010, 184, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Basu, S.; Williams, C.B.; Salzman, N.H.; Dittel, B.N. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J. Immunol. 2012, 188, 3188–3198. [Google Scholar] [CrossRef] [PubMed]
- Ochi, H.; Abraham, M.; Ishikawa, H.; Frenkel, D.; Yang, K.; Basso, A.S.; Wu, H.; Chen, M.L.; Gandhi, R.; Miller, A.; et al. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+ CD25- LAP+ T cells. Nat. Med. 2006, 12, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Yanaba, K.; Bouaziz, J.D.; Fujimoto, M.; Tedder, T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Investig. 2008, 118, 3420–3430. [Google Scholar] [CrossRef] [PubMed]
- Theien, B.E.; Vanderlugt, C.L.; Nickerson-Nutter, C.; Cornebise, M.; Scott, D.M.; Perper, S.J.; Whalley, E.T.; Miller, S.D. Differential effects of treatment with a small-molecule VLA-4 antagonist before and after onset of relapsing EAE. Blood 2003, 102, 4464–4471. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Oshima, H.; Nohara, C.; Morimoto, S.; Yoshino, S.; Kobata, T.; Yagita, H.; Okumura, K. Involvement of CD70–CD27 interactions in the induction of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2000, 109, 188–196. [Google Scholar] [CrossRef]
- MacPhee, I.A.; Turner, D.R.; Yagita, H.; Oliveira, D.B. CD80 (B7.1) and CD86 (B7.2) do not have distinct roles in setting the Th1/Th2 balance in autoimmunity in rats. Scand. J. Immunol. 2001, 54, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Park, I.K.; Hiraki, K.; Ohtani, S.; Kohyama, K. Role of pathogenic T cells and autoantibodies in relapse and progression of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis in LEW.1AV1 rats. Immunology 2009, 128, e250–e261. [Google Scholar] [CrossRef] [PubMed]
- Sefia, E.; Pryce, G.; Meier, U.C.; Giovannoni, G.; Baker, D. Depletion of CD20 B cells fails to inhibit relapsing mouse experimental autoimmune encephalomyelitis. Mult. Scler. Relat. Disorders 2017, 14, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.R. Anti-inflammatory immunotherapy for multiple sclerosis/experimental autoimmune encephalomyelitis (EAE) disease. Curr. Med. Chem. 2005, 12, 2947–2962. [Google Scholar] [CrossRef] [PubMed]
- Theien, B.E.; Vanderlugt, C.L.; Eagar, T.N.; Nickerson-Nutter, C.; Nazareno, R.; Kuchroo, V.K.; Miller, S.D. Discordant effects of anti-VLA-4 treatment before and after onset of relapsing experimental autoimmune encephalomyelitis. J. Clin. Investig. 2001, 107, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Hausler, D.; Nessler, S.; Kruse, N.; Bruck, W.; Metz, I. Natalizumab analogon therapy is effective in a B cell-dependent multiple sclerosis model. Neuropathol. Appl. Neurobiol. 2015, 41, 814–831. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; Rahgozar, K.; Hallworth, N.; Lanker, S.; Carrithers, M.D. Epithelial V-like antigen mediates efficacy of anti-α4 integrin treatment in a mouse model of multiple sclerosis. PLoS ONE 2013, 8, e70954. [Google Scholar] [CrossRef] [PubMed]
- Stefanich, E.G.; Danilenko, D.M.; Wang, H.; OByrne, S.; Erickson, R.; Gelzleichter, T.; Hiraragi, H.; Chiu, H.; Ivelja, S.; Jeet, S.; et al. A humanized monoclonal antibody targeting the β7 integrin selectively blocks intestinal homing of T lymphocytes. Br. J. Pharmacol. 2011, 162, 1855–1870. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.R.; Harrison, J.E.; Wang, D.; Leung, E.; Mueller, W.; Wagner, N.; Krissansen, G.W. β7 integrins contribute to demyelinating disease of the central nervous system. J. Neuroimmunol. 2000, 103, 146–152. [Google Scholar] [CrossRef]
- Schaub, M.; Issazadeh, S.; Stadlbauer, T.H.; Peach, R.; Sayegh, M.H.; Khoury, S.J. Costimulatory signal blockade in murine relapsing experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1999, 96, 158–166. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Hilliard, B.; Wysocka, M.; Ventura, E.S.; Bhopale, M.K.; Trinchieri, G.; Rostami, A.M. IL-12 reverses the suppressive effect of the CD40 ligand blockade on experimental autoimmune encephalomyelitis (EAE). J. Neurol. Sci. 1999, 171, 60–64. [Google Scholar] [CrossRef]
- Smith, E.S.; Jonason, A.; Reilly, C.; Veeraraghavan, J.; Fisher, T.; Doherty, M.; Klimatcheva, E.; Mallow, C.; Cornelius, C.; Leonard, J.E.; et al. SEMA4D compromises blood-brain barrier, activates microglia, and inhibits remyelination in neurodegenerative disease. Neurobiol. Dis. 2015, 73, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Tischner, D.; Weishaupt, A.; van den Brandt, J.; Muller, N.; Beyersdorf, N.; Ip, C.W.; Toyka, K.V.; Hunig, T.; Gold, R.; Kerkau, T.; et al. Polyclonal expansion of regulatory T cells interferes with effector cell migration in a model of multiple sclerosis. Brain 2006, 129, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Perrin, P.J.; June, C.H.; Maldonado, J.H.; Ratts, R.B.; Racke, M.K. Blockade of CD28 during in vitro activation of encephalitogenic T cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 1999, 163, 1704–1710. [Google Scholar] [PubMed]
- Beyersdorf, N.; Hanke, T.; Kerkau, T.; Hunig, T. CD28 superagonists put a break on autoimmunity by preferentially activating CD4+ CD25+ regulatory T cells. Autoimmu. Rev. 2006, 5, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Pang, P.T.; Chretien, N.; Havari, E.; LaMorte, M.J.; Oliver, J.; Pande, N.; Masterjohn, E.; Carter, K.; Reczek, D.; et al. Reduction of inflammation and preservation of neurological function by anti-CD52 therapy in murine experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2015, 285, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Von Kutzleben, S.; Pryce, G.; Giovannoni, G.; Baker, D. Depletion of CD52 positive cells inhibits the development of CNS autoimmune disease, but deletes an immune-tolerance promoting CD8 T cell population. Implications for secondary autoimmunity of alemtuzumab in multiple sclerosis. Immunology 2017, 150, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Pant, A.B.; Wang, Y.; Mielcarz, D.W.; Kasper, E.J.; Telesford, K.M.; Mishra, M.; Haque, A.; Channon, J.Y.; Kasper, L.H.; Begum-Haque, S. Alteration of CD39+ Foxp3+ CD4 T cell and cytokine levels in EAE/MS following anti-CD52 treatment. J. Neuroimmunol. 2017, 303, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Koh, C.S.; Inoue, A.; Tsuyusaki, J.; Yamazaki, M.; Inaba, Y.; Sekiguchi, Y.; Itoh, M.; Yagita, H.; Komiyama, A. Anti-IL-12 antibody prevents the development and progression of multiple sclerosis-like relapsing—Remitting demyelinating disease in NOD mice induced with myelin oligodendrocyte glycoprotein peptide. J. Neuroimmunol. 2000, 102, 56–66. [Google Scholar] [CrossRef]
- Knier, B.; Rothhammer, V.; Heink, S.; Puk, O.; Graw, J.; Hemmer, B.; Korn, T. Neutralizing IL-17 protects the optic nerve from autoimmune pathology and prevents retinal nerve fiber layer atrophy during experimental autoimmune encephalomyelitis. J. Autoimmun. 2015, 56, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Mangan, P.R.; Su, L.J.; Jenny, V.; Tatum, A.L.; Picarillo, C.; Skala, S.; Ditto, N.; Lin, Z.; Yang, X.; Cotter, P.Z.; et al. Dual inhibition of interleukin-23 and interleukin-17 offers superior efficacy in mouse models of autoimmunity. J. Pharmacol. Exp. Ther. 2015, 354, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Mardiguian, S.; Serres, S.; Ladds, E.; Campbell, S.J.; Wilainam, P.; McFadyen, C.; McAteer, M.; Choudhury, R.P.; Smith, P.; Saunders, F.; et al. Anti-IL-17A treatment reduces clinical score and VCAM-1 expression detected by in vivo magnetic resonance imaging in chronic relapsing EAE ABH mice. Am. J. Pathol. 2013, 182, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Sommereyns, C.; Theate, I.; Michiels, T.; Van Snick, J. Anti-IL-17A autovaccination prevents clinical and histological manifestations of experimental autoimmune encephalomyelitis. Ann. N. Y. Acad. Sci. 2007, 1110, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Nohara, C.; Akiba, H.; Nakajima, A.; Inoue, A.; Koh, C.S.; Ohshima, H.; Yagita, H.; Mizuno, Y.; Okumura, K. Amelioration of experimental autoimmune encephalomyelitis with anti-OX40 ligand monoclonal antibody: A critical role for OX40 ligand in migration, but not development, of pathogenic T cells. J. Immunol. 2001, 166, 2108–2115. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.J.; Myers, K.J.; Dougherty, J.P.; Rosen, H.; Ron, Y. Both anti-CD11a (LFA-1) and anti-CD11b (MAC-1) therapy delay the onset and diminish the severity of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1995, 62, 153–160. [Google Scholar] [CrossRef]
- Sun, J.J.; Ren, Q.G.; Xu, L.; Zhang, Z.J. LINGO-1 antibody ameliorates myelin impairment and spatial memory deficits in experimental autoimmune encephalomyelitis mice. Sci. Rep. 2015, 5, 14235. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Spittle, E.; Backstrom, B.T. Partial depletion of CD69low-expressing natural regulatory T cells with the anti-CD25 monoclonal antibody PC61. Scand. J. Immunol. 2007, 65, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Fazekas, G.; Hara, H.; Tabira, T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 163, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Leavenworth, J.W.; Schellack, C.; Kim, H.J.; Lu, L.; Spee, P.; Cantor, H. Analysis of the cellular mechanism underlying inhibition of EAE after treatment with anti-NKG2A F(ab’)2. Proc. Natl. Acad. Sci. USA 2010, 107, 2562–2567. [Google Scholar] [CrossRef] [PubMed]
- Kuchroo, V.K.; Das, M.P.; Brown, J.A.; Ranger, A.M.; Zamvil, S.S.; Sobel, R.A.; Weiner, H.L.; Nabavi, N.; Glimcher, L.H. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: Application to autoimmune disease therapy. Cell 1995, 80, 707–718. [Google Scholar] [CrossRef]
- Karandikar, N.J.; Eagar, T.N.; Vanderlugt, C.L.; Bluestone, J.A.; Miller, S.D. CTLA-4 downregulates epitope spreading and mediates remission in relapsing experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2000, 109, 173–180. [Google Scholar] [CrossRef]
- Heremans, H.; Dillen, C.; Groenen, M.; Martens, E.; Billiau, A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon- γ. Eur. Ournal Immunol. 1996, 26, 2393–2398. [Google Scholar] [CrossRef] [PubMed]
- Voorthuis, J.A.; Uitdehaag, B.M.; de Groot, C.J.; Goede, P.H.; van der Meide, P.H.; Dijkstra, C.D. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-γ in Lewis rats. Clin. Exp.Immunol. 1990, 81, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Najafian, N.; Reddy, J.; Albin, M.; Zhu, C.; Jensen, E.; Imitola, J.; Korn, T.; Anderson, A.C.; Zhang, Z.; et al. Differential engagement of Tim-1 during activation can positively or negatively costimulate T cell expansion and effector function. J. Exp. Med. 2007, 204, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Buonsanti, C.; Mariani, M.; Cella, M.; Gilfillan, S.; Cross, A.H.; Colonna, M.; Panina-Bordignon, P. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2007, 37, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Buonsanti, C.; Cella, M.; Tassi, I.; Schmidt, R.E.; Fenoglio, C.; Rinker, J., 2nd; Naismith, R.T.; Panina-Bordignon, P.; Passini, N.; et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 2008, 131, 3081–3091. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lin, X.; Chen, H.M.; Wu, Q.; Subudhi, S.K.; Chen, L.; Fu, Y.X. Administration of agonistic anti-4–1BB monoclonal antibody leads to the amelioration of experimental autoimmune encephalomyelitis. J. Immunol. 2002, 168, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Sporici, R.; Issekutz, T.B. CXCR3 blockade inhibits T-cell migration into the CNS during EAE and prevents development of adoptively transferred, but not actively induced, disease. Eur. J. Immunol. 2010, 40, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.; Barthelmes, J.; Stolz, L.; Beyer, S.; Diehl, O.; Tegeder, I. “Disease modifying nutricals” for multiple sclerosis. Pharmacol. Ther. 2015, 148, 85–113. [Google Scholar] [CrossRef] [PubMed]
- Brod, S.A.; Bauer, V.L. Ingested (oral) tocilizumab inhibits EAE. Cytokine 2014, 68, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ireland, S.J.; Davis, L.S.; Kong, X.; Stowe, A.M.; Wang, Y.; White, W.I.; Herbst, R.; Monson, N.L. Autoreactive CD19+CD20- plasma cells contribute to disease severity of experimental autoimmune encephalomyelitis. J. Immunol. 2016, 196, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Horikawa, M.; Iwata, Y.; Tedder, T.F. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J. Immunol. 2010, 185, 2240–2252. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitz, K.; Geisslinger, G.; Tegeder, I. Monoclonal Antibodies in Preclinical EAE Models of Multiple Sclerosis: A Systematic Review. Int. J. Mol. Sci. 2017, 18, 1992. https://doi.org/10.3390/ijms18091992
Schmitz K, Geisslinger G, Tegeder I. Monoclonal Antibodies in Preclinical EAE Models of Multiple Sclerosis: A Systematic Review. International Journal of Molecular Sciences. 2017; 18(9):1992. https://doi.org/10.3390/ijms18091992
Chicago/Turabian StyleSchmitz, Katja, Gerd Geisslinger, and Irmgard Tegeder. 2017. "Monoclonal Antibodies in Preclinical EAE Models of Multiple Sclerosis: A Systematic Review" International Journal of Molecular Sciences 18, no. 9: 1992. https://doi.org/10.3390/ijms18091992
APA StyleSchmitz, K., Geisslinger, G., & Tegeder, I. (2017). Monoclonal Antibodies in Preclinical EAE Models of Multiple Sclerosis: A Systematic Review. International Journal of Molecular Sciences, 18(9), 1992. https://doi.org/10.3390/ijms18091992