Individualizing Treatment Approaches for Epileptic Patients with Glucose Transporter Type1 (GLUT-1) Deficiency

Abstract
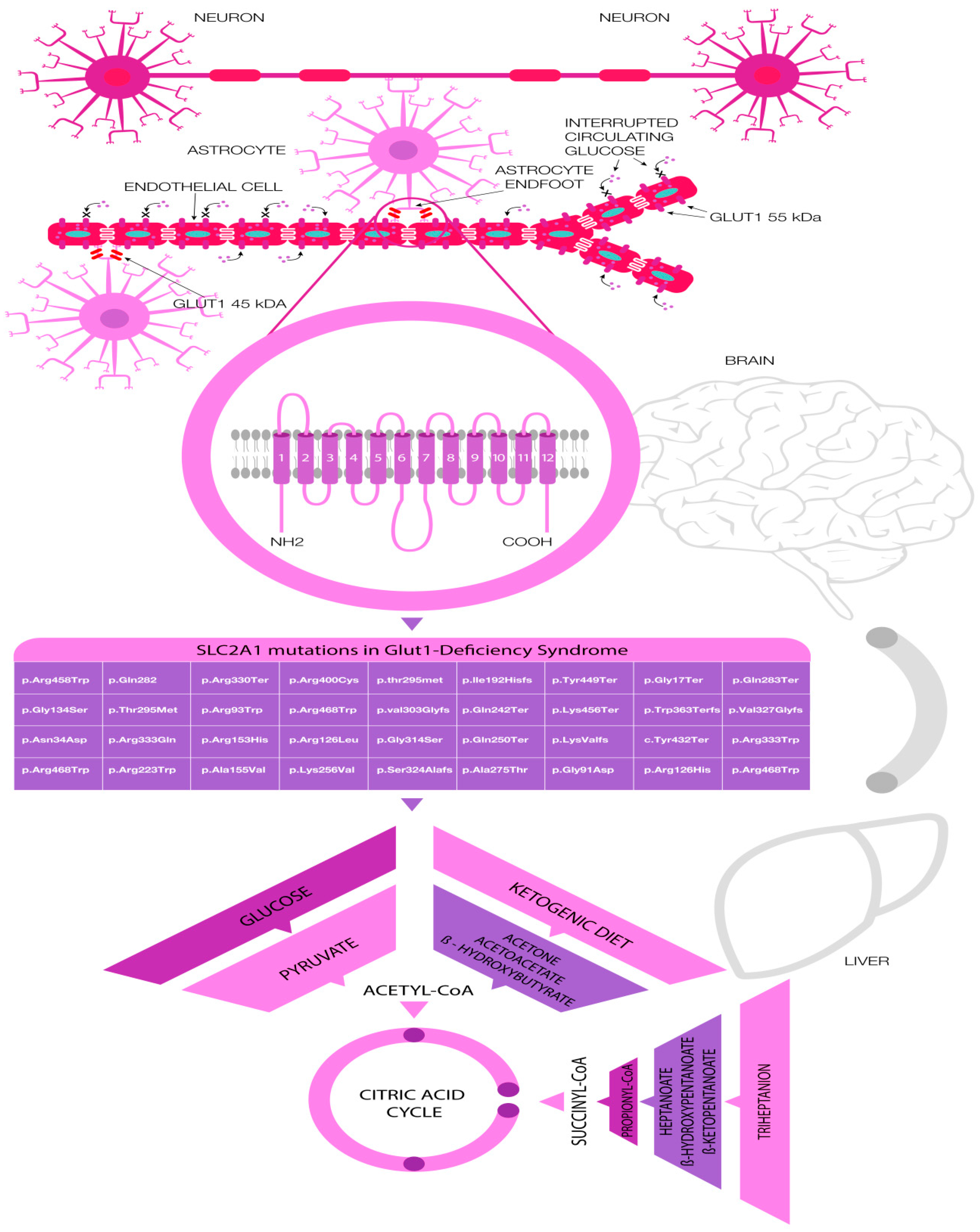
:1. Introduction
2. Glut1 Deficiency Syndrome Treatment Individualizing Approaches
3. Genetic Testing in Glut1-DS
4. Common Pathogenic Variants in GLUT-1 Deficiency
5. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fisher, R.S.; Van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J. Epileptic seizures and epilepsy: Definitions proposed by the International League against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Thurman, D.J.; Beghi, E.; Begley, C.E.; Berg, A.T.; Buchhalter, J.R.; Ding, D.; Hesdorffer, D.C.; Hauser, W.A.; Kazis, L.; Kobau, R.; et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 2011, 52, 2–26. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E. Mechanisms of action of antiepileptic drugs: The search for synergy. Curr. Opin. Neurol. 2010, 23, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Schmidt, D. Epilepsy: Perampanel—New promise for refractory epilepsy? Nat. Rev. Neurol. 2012, 8, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Amalou, S.; Gras, D.; Ilea, A.; Greneche, M.-O.; Francois, L.; Bellavoine, V.; Delanoe, C.; Auvin, S. Use of modified Atkins diet in glucose transporter type 1 deficiency syndrome. Dev. Med. Child Neurol. 2016, 58, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Oguni, H.; Ito, S.; Oguni, M.; Osawa, M. A modified Atkins diet is promising as a treatment for glucose transporter type 1 deficiency syndrome. Dev. Med. Child Neurol. 2011, 53, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Veggiotti, P.; De Giorgis, V. Dietary treatments and new therapeutic perspective in GLUT1 deficiency syndrome. Curr. Treat. Options Neurol. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- De Giorgis, V.; Veggiotti, P. GLUT1 deficiency syndrome 2013: Current state of the art. Seizure 2013, 22, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Franco, V.; Perucca, E. The pharmacogenomics of epilepsy. Expert Rev. Neurother. 2015, 15, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-Resistant Epilepsy. N. Engl. J. Med. 2011, 365, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Cavalleri, G.L.; McCormack, M.; Alhusaini, S.; Chaila, E.; Delanty, N. Pharmacogenomics and epilepsy: The road ahead. Pharmacogenomics 2011, 12, 1429–1447. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.E.; Mirza, N.; Yip, V.L.M.; Marson, A.G.; Pirmohamed, M. Personalized medicine approaches in epilepsy. J. Intern. Med. 2015, 277, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, S.; Sisodiya, S.M. Pharmacogenomics in epilepsy. Neurosci. Lett. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hodson, R. Precision medicine. Nature 2016, 537, S49. [Google Scholar] [CrossRef] [PubMed]
- Reif, P.S.; Tsai, M.H.; Helbig, I.; Rosenow, F.; Klein, K.M. Precision medicine in genetic epilepsies: Break of dawn? Expert Rev. Neurother. 2017, 17, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Gao, G.; Rueda, C.B.; Yu, H.; Thibodeaux, D.N.; Awano, T.; Engelstad, K.M.; Sanchez-Quintero, M.J.; Yang, H.; Li, F.; et al. Brain microvasculature defects and Glut1 deficiency syndrome averted by early repletion of the glucose transporter-1 protein. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Sands, T.T.; Choi, H. Genetic Testing in Pediatric Epilepsy. Curr. Neurol. Neurosci. Rep. 2017, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Weber, Y.G.; Biskup, S.; Helbig, K.L.; Von Spiczak, S.; Lerche, H. The role of genetic testing in epilepsy diagnosis and management. Expert Rev. Mol. Diagn. 2017, 17, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Weber, Y.G.; Nies, A.T.; Schwab, M.; Lerche, H. Genetic Biomarkers in Epilepsy. Neurotherapeutics 2014, 11, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Bizec, C.L.; Nicole, S.; Panagiotakaki, E.; Seta, N.; Vuillaumier-Barrot, S. No Mutation in the SLC2A3 Gene in Cohorts of GLUT1 Deficiency Syndrome-Like Patients Negative for SLC2A1 and in Patients with AHC Negative for ATP1A3. JIMD Rep. 2014, 12, 115–120. [Google Scholar] [PubMed]
- Delanty, N.; Cavallleri, G. Genomics-Guided Precise Anti-Epileptic Drug Development. Neurochem. Res. 2017, 42, 2084–2088. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.; Johannesen, K.M.; Ek, J.; Tang, S.; Marini, C.; Blichfeldt, S.; Kibæk, M.; Von Spiczak, S.; Weckhuysen, S.; Frangu, M.; et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia 2015, 56, e203–e208. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, M.; Cirillo, M.L. Understanding the Spectrum of SLC2A1-Associated Disorders. Pediatr. Neurobiol. Briefs 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Lebon, S.; Suarez, P.; Alija, S.; Korff, C.M.; Fluss, J.; Mercati, D.; Datta, A.N.; Poloni, C.; Marcoz, J.P.; Campos-Xavier, A.B.; et al. When should clinicians search for GLUT1 deficiency syndrome in childhood generalized epilepsies? Eur. J. Paediatr. Neurol. 2015, 19, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.S.; Damiano, J.A.; Mullen, S.A.; Bellows, S.T.; Oliver, K.L.; Dahl, H.H.M.; Scheffer, I.E.; Berkovic, S.F. Glucose metabolism transporters and epilepsy: Only GLUT1 has an established role. Epilepsia 2014, 55. [Google Scholar] [CrossRef] [PubMed]
- Mei, D.; Parrini, E.; Marini, C.; Guerrini, R. The Impact of Next-Generation Sequencing on the Diagnosis and Treatment of Epilepsy in Paediatric Patients. Mol. Diagn. Ther. 2017, 21, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.A.; Mullen, S.; Kim, T.H.; Petrou, S. Epilepsy, energy deficiency and new therapeutic approaches including diet. Pharmacol. Ther. 2014, 144, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Borges, K.; Petrou, S.; Reid, C.A. Triheptanoin reduces seizure susceptibility in a syndrome-specific mouse model of generalized epilepsy. Epilepsy Res. 2013, 103, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Burton, B.; Berry, G.T.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; Humphrey, R.; et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: Safety and efficacy in children and adults following 24 weeks of treatment. Mol. Genet. Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Hainque, E.; Gras, D.; Adanyeguh, I.M.; Caillet, S.; Héron, B.; Roubertie, A.; Kaphan, E.; Valabregue, R.; Rinaldi, D.; et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J. Neurol. Neurosurg. Psychiatry 2016, 87, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.M.; Liu, P.; Mao, D.; Kelly, D.I.; Hernandez, A.; Sheng, M.; Good, L.B.; Ma, Q.; Marin-Valencia, I.; Zhang, X.; et al. Triheptanoin for glucose transporter type 1 deficiency (G1D): Modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. 2014, 71, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Abramov, A.Y.; Walker, M.C. Energy depletion in seizures: Anaplerosis as a strategy for future therapies. Neuropharmacology 2013, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F. Triheptanoin for the treatment of brain energy deficit: A 14-year experience. J. Neurosci. Res. 2017, 95, 2236–2243. [Google Scholar] [CrossRef] [PubMed]
- Tzadok, M.; Nissenkorn, A.; Porper, K.; Matot, I.; Marcu, S.; Anikster, Y.; Menascu, S.; Bercovich, D.; Ben Zeev, B. The many faces of Glut1 deficiency syndrome. J. Child Neurol. 2014, 29, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Pong, A.W.; Geary, B.R.; Engelstad, K.M.; Natarajan, A.; Yang, H.; De Vivo, D.C. Glucose transporter type 1 deficiency syndrome: Epilepsy phenotypes and outcomes. Epilepsia 2012, 53, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pascual, J.M.; De Vivo, D. Glucose Transporter Type 1 Deficiency Syndrome; University of Washington: Seattle, WA, USA, 2015. [Google Scholar]
- Klepper, J.; Flörcken, A.; Fischbarg, J.; Voit, T. Effects of anticonvulsants on GLUT1-mediated glucose transport in GLUT1 deficiency syndrome in vitro. Eur. J. Pediatr. 2003, 162, 84–89. [Google Scholar] [PubMed]
- Kass, H.R.; Winesett, S.P.; Bessone, S.K.; Turner, Z.; Kossoff, E.H. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure 2016, 35, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Osaka, H.; Muramatsu, S.; Takino, N.; Ito, M.; Aoki, S.; Jimbo, E.F.; Shimazaki, K.; Onaka, T.; Ohtsuki, S.; et al. Gene therapy for a mouse model of glucose transporter-1 deficiency syndrome. Mol. Genet. Metab. Rep. 2017, 10, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Weber, Y.G.; Toliat, M.R.; Schubert, J.; Leu, C.; Chaimana, R.; Baulac, S.; Guerrero, R.; LeGuern, E.; Lehesjoki, A.E.; et al. GLUT1 mutations are a rare cause of familial idiopathic generalized epilepsy. Neurology 2012, 78, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Kagitani-Shimono, K.; Sakai, N.; Otomo, T.; Tominaga, K.; Nabatame, S.; Mogami, Y.; Takahashi, Y.; Imai, K.; Yanagihara, K.; et al. SLC2A1 gene analysis of Japanese patients with glucose transporter 1 deficiency syndrome. J. Hum. Genet. 2011, 56, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Leen, W.G.; Klepper, J.; Verbeek, M.M.; Leferink, M.; Hofste, T.; Van Engelen, B.G.; Wevers, R.A.; Arthur, T.; Bahi-Buisson, N.; Ballhausen, D.; et al. Glucose transporter-1 deficiency syndrome: The expanding clinical and genetic spectrum of a treatable disorder. Brain 2010, 133, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.A.S.; Reavey, E. G404(P) Detection rate of slc2a1 gene variants causing glucose transporter 1 deffiency syndrome in patients with childhood onset epilepsy. Arch. Dis. Child. 2017, 159. [Google Scholar] [CrossRef]
- Juozapaite, S.; Praninskiene, R.; Burnyte, B.; Ambrozaityte, L.; Skerliene, B. Novel mutation in a patient with late onset GLUT1 deficiency syndrome. Brain Dev. 2017, 39, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Becker, F.; Schubert, J.; Weckhuysen, S.; Suls, A.; Grüninger, S.; Korn-Merker, E.; Hofmann-Peters, A.; Sperner, J.; Cross, H.; Hallmann, K.; et al. Do Glut1 (glucose transporter type 1) defects exist in epilepsy patients responding to a ketogenic diet? Epilepsy Res. 2015, 114, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Ragona, F.; Matricardi, S.; Castellotti, B.; Patrini, M.; Freri, E.; Binelli, S.; Granata, T. Refractory absence epilepsy and Glut1 deficiency syndrome: A new case report and literature review. Neuropediatrics 2014, 45, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Shimokawa, N.; Hamdan, F.F.; Nassif, C.; Poulin, C.; Al Gazali, L.; Rosenfeld, J.A.; Koibuchi, N.; Rouleau, G.A.; Al Shamsi, A.; et al. Dysfunction of the Cerebral Glucose Transporter SLC45A1 in Individuals with Intellectual Disability and Epilepsy. Am. J. Hum. Genet. 2017, 100, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Çolak, R.; Alkan, Ö.S.; Yangın, E.E.; Kağnıcı, M.; Çalkavur, Ş. A Different SLC2A1 Gene Mutation in Glut 1 Deficiency Syndrome: C.734A>C. Balkan Med. J. 2017, 34, 580–583. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Diet Treatment | Antiepileptic Drugs | Drug Combinations to Avoid with KD | Drugs to Avoid Due to GLUT1 Impairment |
|---|---|---|---|
| Ketogenic (gold standard) | Acetazolamide | Valproate | Phenobarbital, Valproate |
| Modified Atkins | Topiramate | Zonisamide | Tyrosine kinase inhibitors |
| Medium chain Triglycerides | Zonisamide | Acetazolamide | Caffeine, Ethanol |
| Low glycemic index treatment | Phenytoin | Topiramate | Diazepam, Narcotics |
| Triheptanoin (under investigation) | Carbamazepine | - | Tricyclic antidepressants |
| Alpha lipoic acid (under investigation) | - | - | General anaesthetics, Chloral hydrate |
| - | - | - | Guanosine triphosphate analogues |
| DNA Nucleotide Change | Predicted Protein Change | dbSNP | Phenotypes |
|---|---|---|---|
| c.49G>T | p.Gly17Ter | - | NA |
| c.1089delG | p.Trp363Terfs | rs587784391 | A |
| c.1296C>A | p.Tyr432Ter | rs75485205 | A |
| c.1347C>A | p.Tyr449Ter | rs80359828 | A |
| c.1366A>T | p.Lys456Ter | rs80359829 | A |
| c.19_28delAAGCTGACGG | p.Lys7Valfs | rs587784393 | A |
| c.272G>A | p.Gly91Asp | rs80359814 | A |
| c.376C>T | p.Arg126Cys | rs80359818 | A,C |
| c.377G>A | p.Arg126His | rs80359816 | A,C |
| c.574_57delAT | p.lle192Hisfs | rs878853161 | A |
| c.724C>T | p.Gln242Ter | rs794729221 | A |
| c.748C>T | p.Gln250Ter | rs587784396 | NA |
| c.823G>A | p.Ala275Thr | rs121909740 | A,B,C |
| c.847C>T | p.Gln283Ter | rs587784397 | A |
| c.907dupG | p.val303Glyfs | rs7960655334 | A,B |
| c.940G>A | p.Gly314Ser | rs121909739 | A,B |
| c.966_967delCG | p.Ser324Alafs | rs886044287 | NA |
| c.980_981delTG | p.Val327Glyfs | rs80359838 | A,B |
| c.997C>T | p.Arg333Trp | rs80359825 | A,C |
| c.1198C>T | p.Arg400Cys | rs796053263 | NA |
| c.1402C>T | p.Arg468Trp | rs267607059 | A,B |
| c.377G>T | p.Arg126Leu | rs80359816 | A,B |
| c.766_767delAAinsGT | p.Lys256Val | rs80359822 | A |
| c.971C>T | p.Ser324Leu | rs796053253 | B,C |
| c.988C>T | p.Arg330Ter | rs80359826 | A,B,C |
| c.277C>T | p.Arg93Trp | rs267607061 | C |
| c.458G>A | p.Arg153His | rs149585781 | C |
| c.464C>T | p.Ala155Val | rs35313240 | B |
| c.634C>T | p.Arg212Cys | rs61750200 | A,C |
| c.667C>T | p.Arg223Trp | rs757188030 | B,C |
| c.844C>T | p.Gln282 | rs1057521066 | A |
| c.884C>T | p.Thr295Met | rs80359823 | A,C |
| c.998G>A | p.Arg333Gln | rs111033808 | B |
| c.1372C>T | p.Arg458Trp | rs13306758 | A,B |
| c.400G>A | p.Gly134Ser | rs1057518953 | A,C |
| c.100A>G | p.Asn34Asp | rs587784390 | A |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daci, A.; Bozalija, A.; Jashari, F.; Krasniqi, S. Individualizing Treatment Approaches for Epileptic Patients with Glucose Transporter Type1 (GLUT-1) Deficiency. Int. J. Mol. Sci. 2018, 19, 122. https://doi.org/10.3390/ijms19010122
Daci A, Bozalija A, Jashari F, Krasniqi S. Individualizing Treatment Approaches for Epileptic Patients with Glucose Transporter Type1 (GLUT-1) Deficiency. International Journal of Molecular Sciences. 2018; 19(1):122. https://doi.org/10.3390/ijms19010122
Chicago/Turabian StyleDaci, Armond, Adnan Bozalija, Fisnik Jashari, and Shaip Krasniqi. 2018. "Individualizing Treatment Approaches for Epileptic Patients with Glucose Transporter Type1 (GLUT-1) Deficiency" International Journal of Molecular Sciences 19, no. 1: 122. https://doi.org/10.3390/ijms19010122
APA StyleDaci, A., Bozalija, A., Jashari, F., & Krasniqi, S. (2018). Individualizing Treatment Approaches for Epileptic Patients with Glucose Transporter Type1 (GLUT-1) Deficiency. International Journal of Molecular Sciences, 19(1), 122. https://doi.org/10.3390/ijms19010122