Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome



Abstract
:1. Introduction
2. Results and Discussion
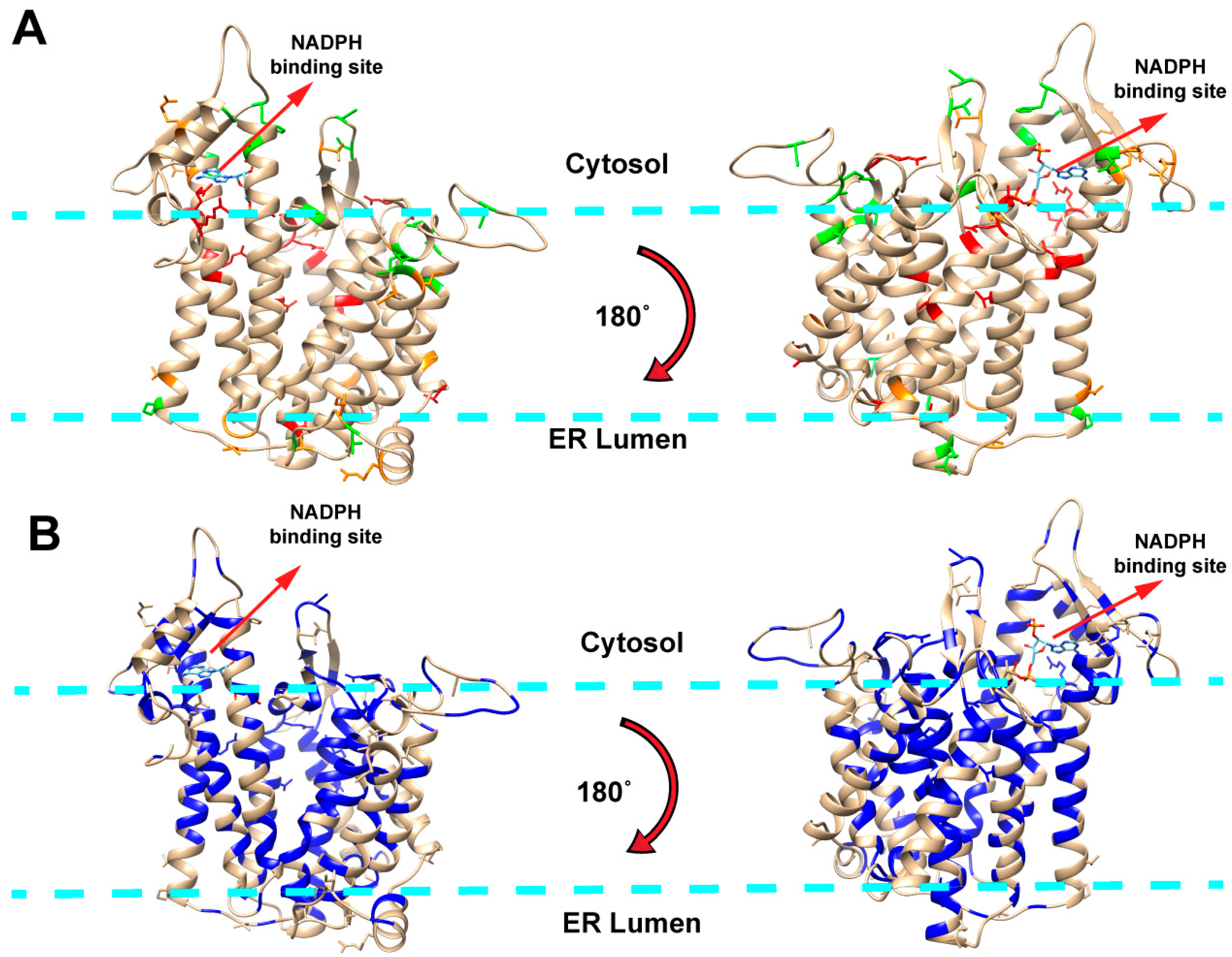
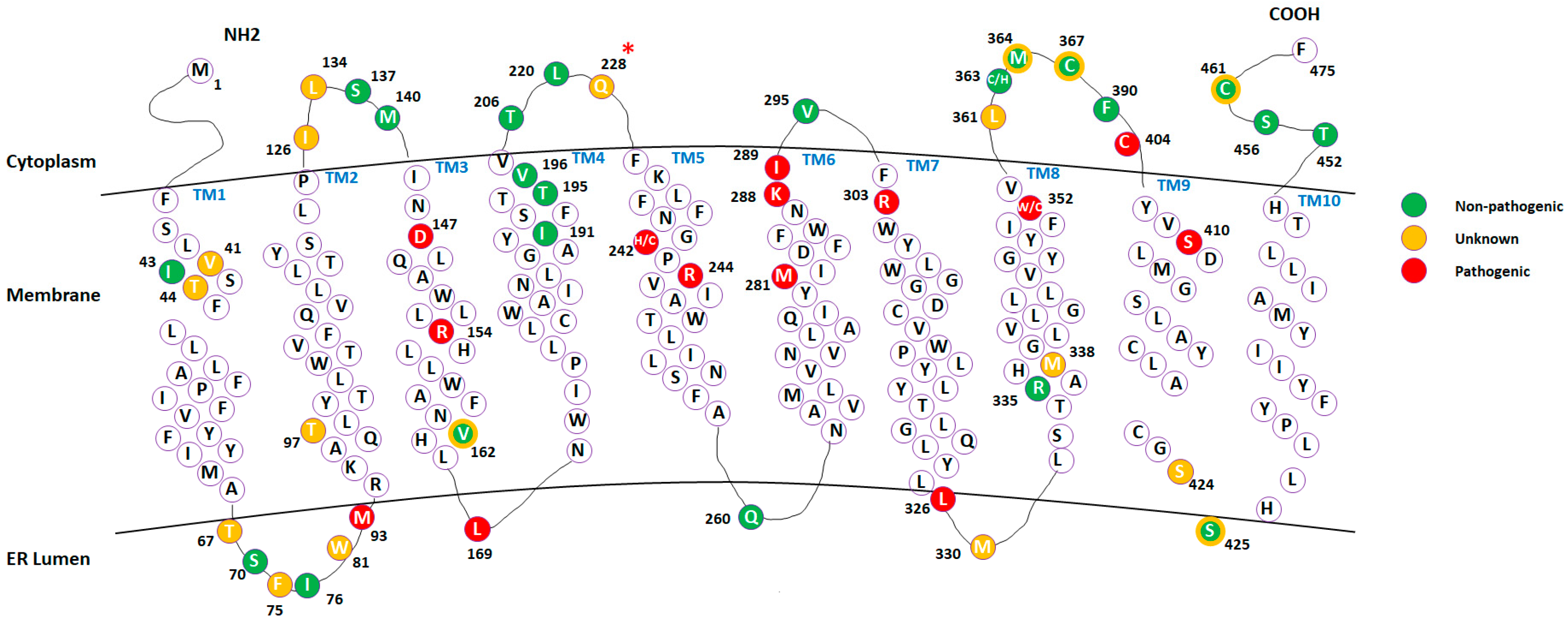
2.1. Mapping Missense Mutations onto the 3D Structure of DHCR7 Protein
2.2. Classification of the Mutations with Unknown Effects Using KNN Model
2.3 Case Study of Selected Mutations Using Molecular Dynamics (MD) Simulations
2.4. Analysis of Mutations’ Pathogenic Effects
2.4.1. Ligand Binding
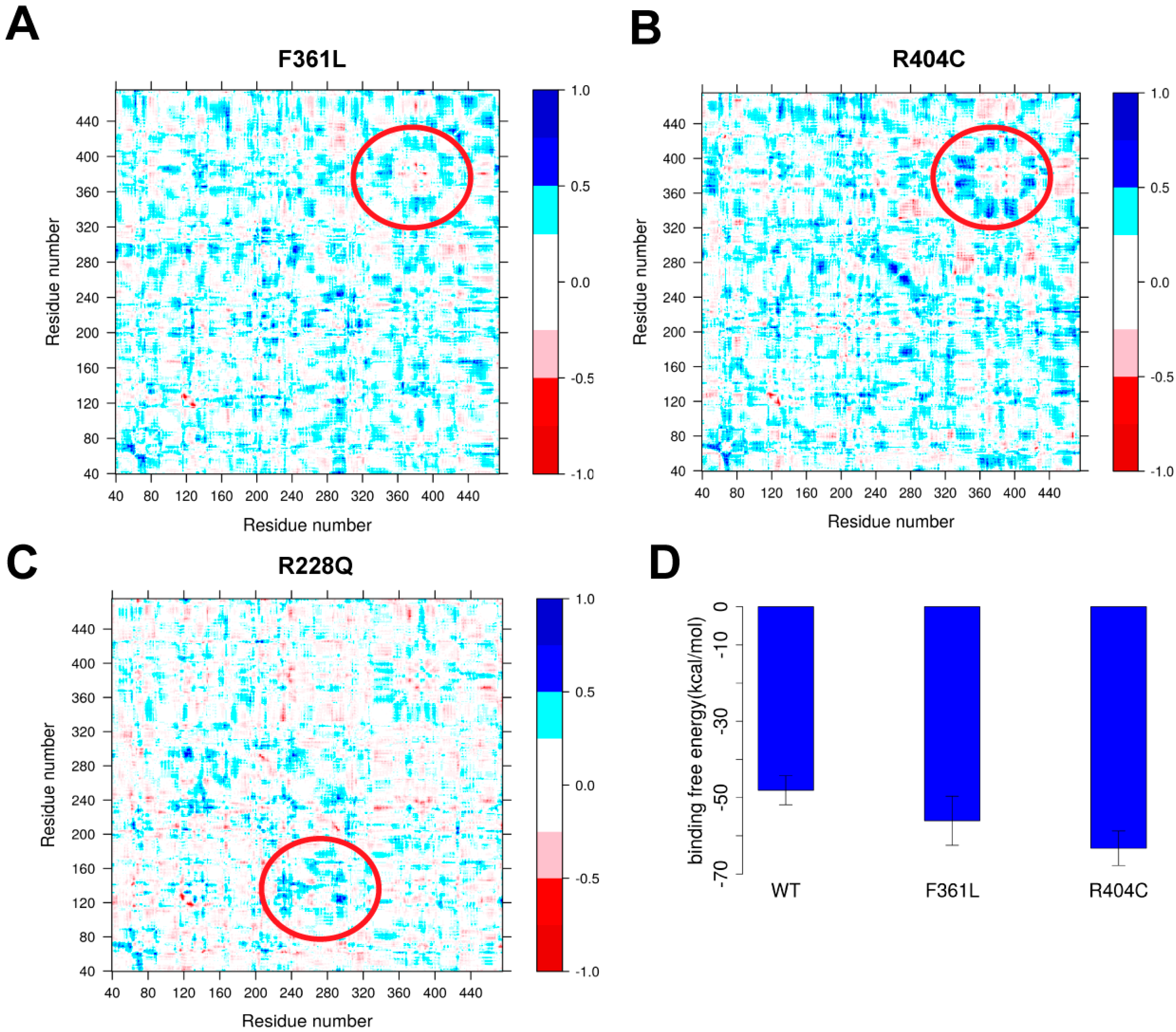
2.4.2. Protein Dynamics
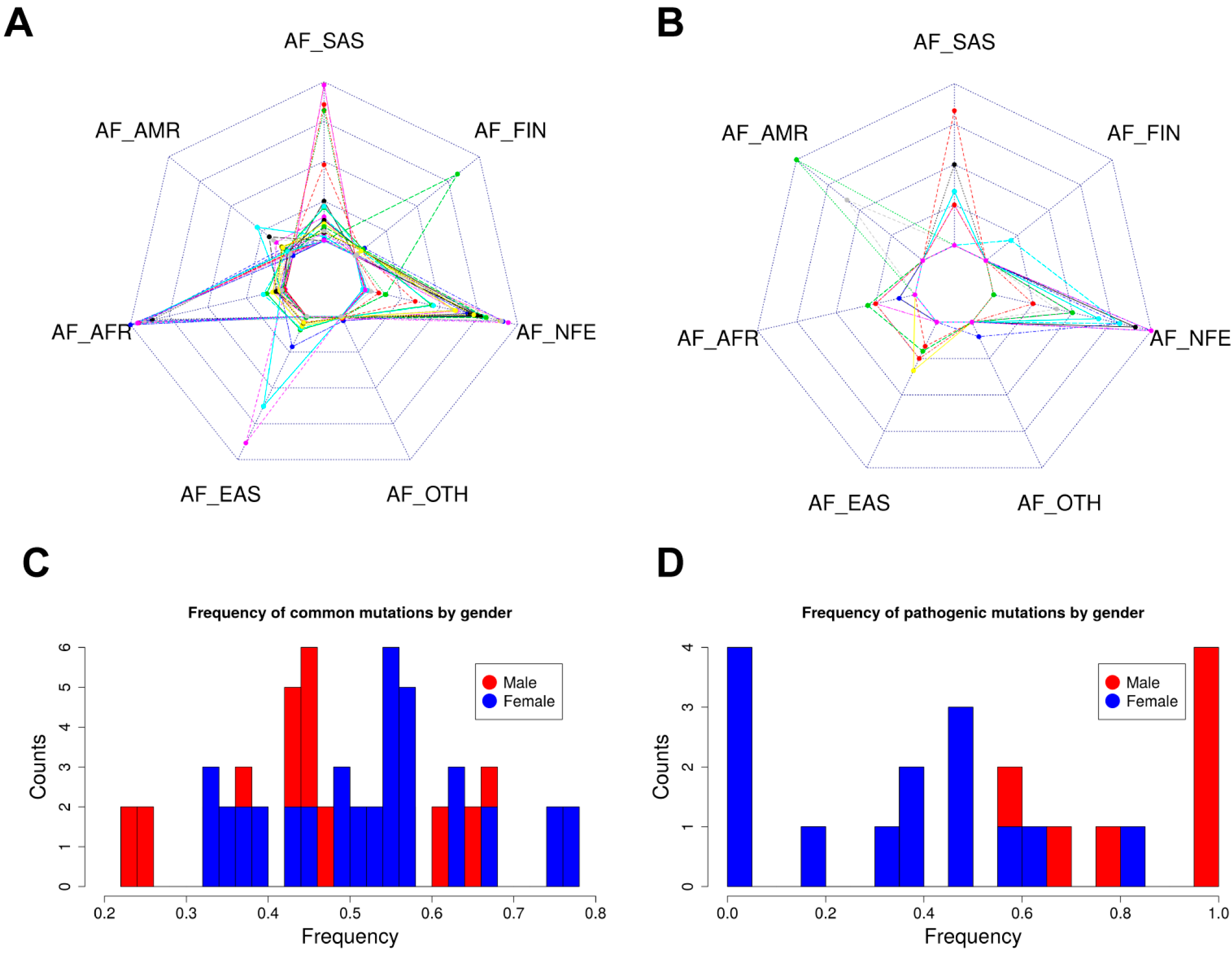
2.5. Allele Frequency Analysis
3. Materials and Methods
3.1. Selection of DHCR7 Missense Variants
3.2. Selection of Non-Pathogenic DHCR7 Mutations
3.3. Obtaining Allele Frequency and Gender Occurrence
3.4. Generation of the 3D Model for DHCR7
3.5. Property Distance (PD)
3.6. Evolutionary Conservation Score (EC Score) Calculation
3.7. Folding Free Energy Change (ΔΔG) and Relative Solvent Accessible Surface Area (rSASA) Calculation
3.8. Molecular Dynamic Simulations
3.9. MM/PBSA Analysis
3.10. K-Nearest Neighbors (KNN) Classification
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
Abbreviations
| SLOS | Smith-Lemli-Opitz syndrome |
| DHCR7 | 7-dehydroxycholesterol reductase |
| ER | Endoplasmic reticulum |
| ExAC | Exome Aggregation Consortium |
| PD | Property distance |
| ECS | Evolutional conservation score |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| SASA | Solvent accessible surface area |
| rSASA | Relative solvent accessible surface area |
| KNN | K-Nearest Neighbors |
| MD | Molecules dynamics |
| CL | Cytosol loops |
| TM | Transmembrane domain |
| ESP | Exome Sequencing Project |
References
- Smith, D.W.; Lemli, L.; Opitz, J.M. A newly recognized syndromeof multiple congenital anomalies. J. Pediatr. 1964, 64, 210–217. [Google Scholar] [CrossRef]
- Battaile, K.P.; Battaile, B.C.; Merkens, L.S.; Maslen, C.L.; Steiner, R.D. Carrier frequency of the common mutation ivs8-1g>c in dhcr7 and estimate of the expected incidence of smith-lemli-opitz syndrome. Mol. Genet. Metab. 2001, 72, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, M.J.; Waye, J.S.; Douketis, J.D. DHCR7 mutation carrier rates and prevalence of the rsh/smith-lemli-opitz syndrome: Where are the patients? Am. J. Med. Genet. Part A 2006, 140, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Tint, G.S.; Salen, G.; Patel, S.B. Detection of a common mutation in the RSH or smith-lemli-opitz syndrome by a PCR-RFLP assay: Ivs8-1g? C is found in over sixty percent of us propositi. Am. J. Med. Genet. Part A 2000, 90, 347–350. [Google Scholar] [CrossRef]
- Waterham, H.R.; Hennekam, R.C. Mutational spectrum of smith-lemli-opitz syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160, 263–284. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I.; Herman, G.E. Inborn errors of sterol biosynthesis. Ann. Rev. Genom. Hum. Genet. 2001, 2, 299–341. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I. The smith-lemli-opitz syndrome. J. Med. Genet. 2000, 37, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.N.; Tuli, S.Y.; Tuli, S.S.; Stern, M.A.; Giordano, B.P. Brothers with smith-lemli-opitz syndrome. J. Pediatr. Health Care 2015, 29, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Witsch-Baumgartner, M.; Fitzky, B.U.; Ogorelkova, M.; Kraft, H.G.; Moebius, F.F.; Glossmann, H.; Seedorf, U.; Gillessen-Kaesbach, G.; Hoffmann, G.F.; Clayton, P.; et al. Mutational spectrum in the delta7-sterol reductase gene and genotype-phenotype correlation in 84 patients with smith-lemli-opitz syndrome. Am. J. Hum. Genet. 2000, 66, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Tint, G.S.; Irons, M.; Elias, E.R.; Batta, A.K.; Frieden, R.; Chen, T.S.; Salen, G. Defective cholesterol biosynthesis associated with the smith-lemli-opitz syndrome. N. Engl. J. Med. 1994, 330, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Shefer, S.; Salen, G.; Batta, A.K.; Honda, A.; Tint, G.S.; Irons, M.; Elias, E.R.; Chen, T.C.; Holick, M.F. Markedly inhibited 7-dehydrocholesterol-delta 7-reductase activity in liver microsomes from smith-lemli-opitz homozygotes. J. Clin. Investig. 1995, 96, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Correa-Cerro, L.S.; Porter, F.D. 3beta-hydroxysterol delta7-reductase and the smith-lemli-opitz syndrome. Mol. Genet. Metab. 2005, 84, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Hossein-nezhad, A.; Holick, M.F. Vitamin d for health: A global perspective. Mayo Clin. Proc. 2013, 88, 720–755. [Google Scholar] [CrossRef] [PubMed]
- Porter, F.D.; Herman, G.E. Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 2011, 52, 6–34. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, A.V.; Luu, W.; Sharpe, L.J.; Brown, A.J. Cholesterol-mediated degradation of 7-dehydrocholesterol reductase switches the balance from cholesterol to vitamin d synthesis. J. Biol. Chem. 2016, 291, 8363–8373. [Google Scholar] [CrossRef] [PubMed]
- Kuan, V.; Martineau, A.R.; Griffiths, C.J.; Hypponen, E.; Walton, R. Dhcr7 mutations linked to higher vitamin d status allowed early human migration to northern latitudes. BMC Evol. Biol. 2013, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Moebius, F.F.; Fitzky, B.U.; Lee, J.N.; Paik, Y.K.; Glossmann, H. Molecular cloning and expression of the human delta7-sterol reductase. Proc. Natl. Acad. Sci. USA 1998, 95, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Fitzky, B.U.; Witsch-Baumgartner, M.; Erdel, M.; Lee, J.N.; Paik, Y.K.; Glossmann, H.; Utermann, G.; Moebius, F.F. Mutations in the delta7-sterol reductase gene in patients with the smith-lemli-opitz syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 8181–8186. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Roberti, R.; Blobel, G. Structure of an integral membrane sterol reductase from methylomicrobium alcaliphilum. Nature 2015, 517, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Kroncke, B.M.; Duran, A.M.; Mendenhall, J.L.; Meiler, J.; Blume, J.D.; Sanders, C.R. Documentation of an imperative to improve methods for predicting membrane protein stability. Biochemistry 2016, 55, 5002–5009. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nssnps on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Norris, J.; Schwartz, C.; Alexov, E. Revealing the effects of missense mutations causing snyder-robinson syndrome on the stability and dimerization of spermine synthase. Int. J. Mol. Sci. 2016, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Suryadi, J.; Yang, Y.; Kucukkal, T.G.; Cao, W.; Alexov, E. Mutations in the kdm5c arid domain and their plausible association with syndromic claes-jensen-type disease. Int. J. Mol. Sci. 2015, 16, 27270–27287. [Google Scholar] [CrossRef] [PubMed]
- Cubellis, M.V.; Baaden, M.; Andreotti, G. Taming molecular flexibility to tackle rare diseases. Biochimie 2015, 113, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, A.V.; Luu, W.; Li, D.; Sharpe, L.J.; Brown, A.J. Dhcr7: A vital enzyme switch between cholesterol and vitamin d production. Prog. Lipid Res. 2016, 64, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An r package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- Estacio, S.G.; Shakhnovich, E.I.; Faisca, P.F. Assessing the effect of loop mutations in the folding space of beta2-microglobulin with molecular dynamics simulations. Int. J. Mol. Sci. 2013, 14, 17256–17278. [Google Scholar] [CrossRef] [PubMed]
- Witham, S.; Takano, K.; Schwartz, C.; Alexov, E. A missense mutation in clic2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins 2011, 79, 2444–2454. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Alexov, E. Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins 2016, 84, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Witsch-Baumgartner, M.; Schwentner, I.; Gruber, M.; Benlian, P.; Bertranpetit, J.; Bieth, E.; Chevy, F.; Clusellas, N.; Estivill, X.; Gasparini, G.; et al. Age and origin of major smith-lemli-opitz syndrome (SLOS) mutations in european populations. J. Med. Genet. 2008, 45, 200–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. Clinvar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Romano, F.; Fiore, B.; Pezzino, F.M.; Longombardo, M.T.; Cefalù, A.B.; Noto, D.; Puglisi, A.; Brogna, A.; Mattina, T.; Averna, M.; et al. A novel mutation of the dhcr7 gene in a sicilian compound heterozygote with smith-lemli-opitz syndrome. Mol. Diagn. 2012, 9, 201–204. [Google Scholar] [CrossRef]
- Tamura, M.; Isojima, T.; Kasama, T.; Mafune, R.; Shimoda, K.; Yasudo, H.; Tanaka, H.; Takahashi, C.; Oka, A.; Kitanaka, S. Novel dhcr7 mutation in a case of smith-lemli-opitz syndrome showing 46,xy disorder of sex development. Hum. Genome Var. 2017, 4, 17015. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using modeller. Curr. Protoc. Bioinform. 2014, 47, 5–6. [Google Scholar]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Sun, L.; Jia, Z.; Li, L.; Alexov, E. Predicting protein-DNA binding free energy change upon missense mutations using modified mm/pbsa approach: Sampdi webserver. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. Uniprot: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. Duet: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Ding, F.; Dokholyan, N.V. Eris: An automated estimator of protein stability. Nat. Methods 2007, 4, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. Mcsm: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Topham, C.M.; Srinivasan, N.; Blundell, T.L. Prediction of the stability of protein mutants based on structural environment-dependent amino acid substitution and propensity tables. Protein Eng. Des. Sel. 1997, 10, 7–21. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The foldx web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Getov, I.; Petukh, M.; Alexov, E. Saafec: Predicting the effect of single point mutations on protein folding free energy using a knowledge-modified mm/pbsa approach. Int. J. Mol. Sci. 2016, 17, 512. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [PubMed]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. Opm: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with namd. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, C.; Sarkar, S.; Zhang, J.; Witham, S.; Zhang, Z.; Wang, L.; Smith, N.; Petukh, M.; Alexov, E. Delphi: A comprehensive suite for delphi software and associated resources. BMC Biophys. 2012, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Estacio, S.G.; Martiniano, H.F.; Faisca, P.F. Thermal unfolding simulations of nbd1 domain variants reveal structural motifs associated with the impaired folding of f508del-cftr. Mol. Biosyst. 2016, 12, 2834–2848. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | KNN Classification | Polyphen | Mutation | KNN Classification | Polyphen |
|---|---|---|---|---|---|
| A41V | N | Benign | R228Q | P | Probably damaging |
| I44T | N | Benign | V330M | N | Probably damaging |
| A67T | N | Possibly damaging | V338M | N | Benign |
| I75F | N | Benign | F361L | N | Probably damaging |
| R81W | N | Probably damaging | T364M | N | Probably damaging |
| A97T | N | Possibly damaging | R367C | N | Probably damaging |
| V126I | N | Probably damaging | G424S | N | Probably damaging |
| V134L | N | Benign | G425S | N | Benign |
| A162V | N | Possibly damaging | R461C | N | Probably damaging |
| Pathogenic Missense Mutations | |||||||||||||||
| TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | TM8 | TM9_10 | CL1 | CL2 | CL3 | CL4 | CTD | TM1+TM2-TM7-TM9_10+CL2 | |
| T154R | 22.9 | 17.6 | 15.5 | 9.1 | 14.5 | 17.4 | 17.2 | 17.8 | 31.6 | 53.1 | 48.6 | 8.8 | 78.8 | 30.8 | 40.3 |
| E288K | 19.6 | 16.4 | 16.0 | 10.5 | 13.1 | 14.3 | 16.0 | 18.4 | 26.1 | 49.2 | 38.6 | 13.6 | 77.5 | 32.0 | 32.7 |
| T289I | 25.2 | 18.0 | 17.8 | 12.8 | 14.1 | 14.3 | 19.5 | 17.5 | 28.7 | 50.1 | 47.9 | 10.1 | 71.1 | 30.4 | 42.9 |
| G303R | 21.1 | 18.9 | 16.8 | 11.0 | 13.3 | 16.0 | 18.2 | 17.0 | 30.2 | 50.5 | 49.2 | 10.4 | 65.0 | 30.1 | 40.9 |
| R404C | 23.4 | 16.5 | 16.0 | 10.8 | 16.0 | 16.8 | 20.8 | 23.3 | 31.6 | 48.9 | 57.4 | 10.0 | 80.1 | 32.7 | 44.9 |
| Missense Mutations with Unknown Effects | |||||||||||||||
| TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | TM8 | TM9_10 | CL1 | CL2 | CL3 | CL4 | CTD | TM1+TM2-TM7-TM9_10+CL2 | |
| V134L | 20.1 | 21.1 | 18.8 | 11.0 | 14.7 | 13.6 | 16.6 | 16.2 | 27.7 | 52.9 | 53.4 | 11.0 | 79.7 | 35.6 | 50.3 |
| R228Q | 17.6 | 17.0 | 15.9 | 8.5 | 13.6 | 13.0 | 15.6 | 16.9 | 27.6 | 53.2 | 54.2 | 10.7 | 75.8 | 36.7 | 45.6 |
| F361L | 19.4 | 17.4 | 14.8 | 9.9 | 14.2 | 14.0 | 18.3 | 16.6 | 28.8 | 54.7 | 50.8 | 11.7 | 74.8 | 33.5 | 40.5 |
| Non-Pathogenic Missense Mutations | |||||||||||||||
| TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | TM8 | TM9_10 | CL1 | CL2 | CL3 | CL4 | CTD | TM1+TM2-TM7-TM9_10+CL2 | |
| R260Q | 19.7 | 18.6 | 15.5 | 9.4 | 12.9 | 14.6 | 15.4 | 17.2 | 24.4 | 58.4 | 52.1 | 11.4 | 79.3 | 28.1 | 50.6 |
| A452T | 20.9 | 19.6 | 17.8 | 10.5 | 13.6 | 16.2 | 16.4 | 17.2 | 26.0 | 55.2 | 52.8 | 8.7 | 66.6 | 30.1 | 51.0 |
| Wild Type | |||||||||||||||
| TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | TM8 | TM9_10 | CL1 | CL2 | CL3 | CL4 | CTD | TM1+TM2-TM7-TM9_10+CL2 | |
| WT | 18.2 | 18.3 | 17.9 | 10.7 | 16.3 | 16.0 | 18.5 | 18.1 | 31.1 | 51.9 | 65.1 | 13.0 | 80.4 | 37.8 | 52.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Myers, R.; Zhang, W.; Alexov, E. Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome. Int. J. Mol. Sci. 2018, 19, 141. https://doi.org/10.3390/ijms19010141
Peng Y, Myers R, Zhang W, Alexov E. Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome. International Journal of Molecular Sciences. 2018; 19(1):141. https://doi.org/10.3390/ijms19010141
Chicago/Turabian StylePeng, Yunhui, Rebecca Myers, Wenxing Zhang, and Emil Alexov. 2018. "Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome" International Journal of Molecular Sciences 19, no. 1: 141. https://doi.org/10.3390/ijms19010141
APA StylePeng, Y., Myers, R., Zhang, W., & Alexov, E. (2018). Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome. International Journal of Molecular Sciences, 19(1), 141. https://doi.org/10.3390/ijms19010141