Psoriasis: A STAT3-Centric View


Abstract
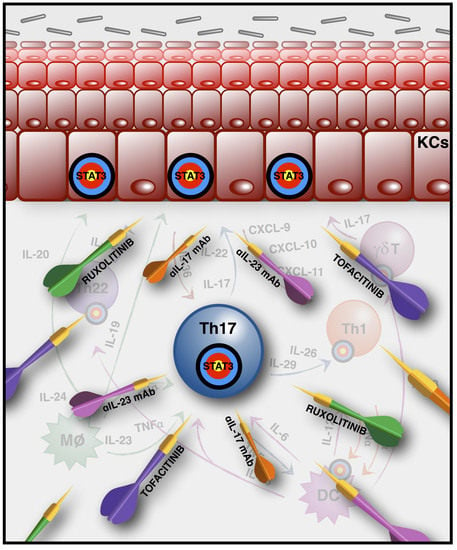
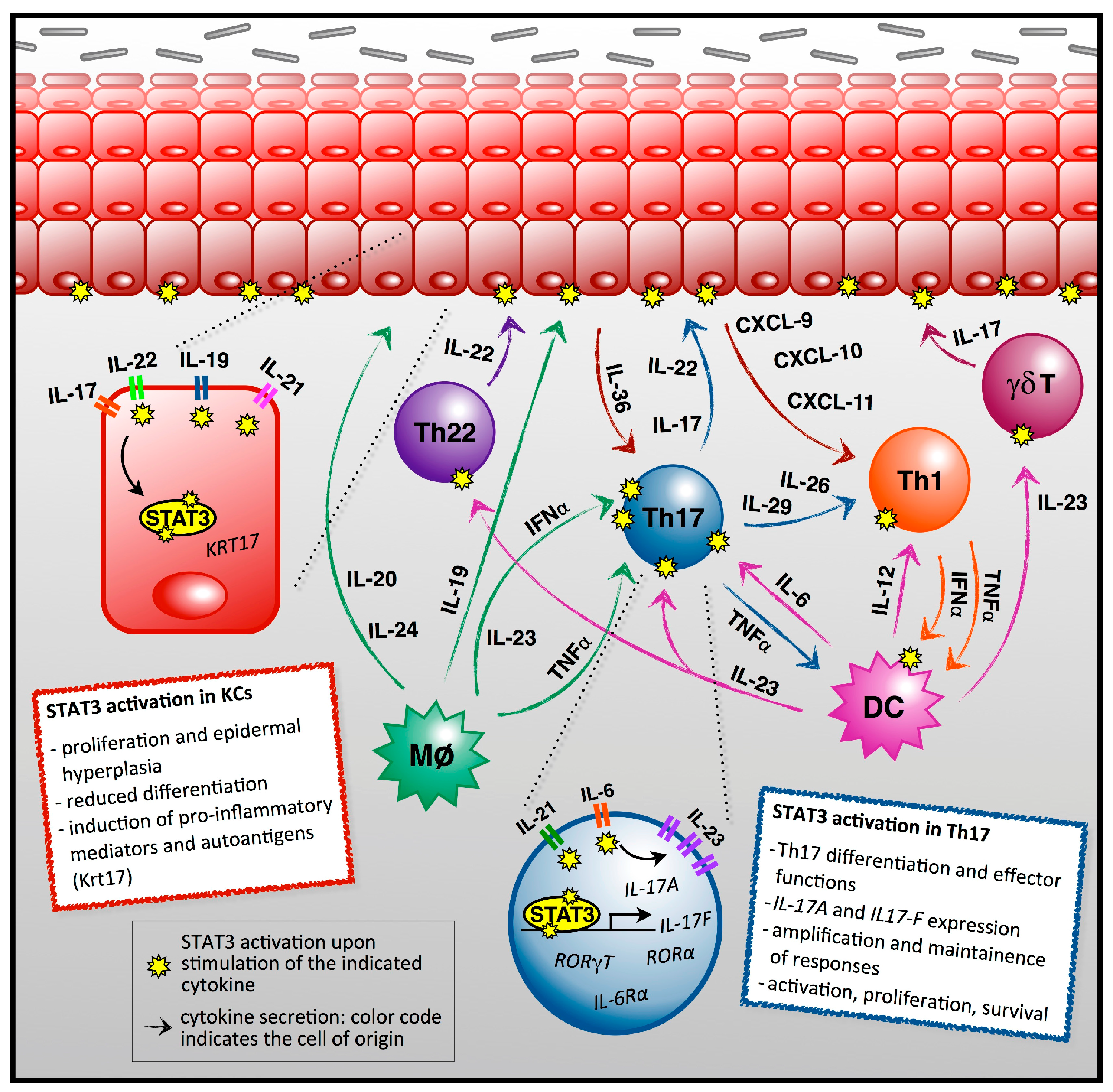
:
1. The Main Players in Psoriasis Pathogenesis
2. STAT3 as a Central Player in Psoriasis Pathogenesis
3. Biological Therapeutic Strategies (Anti-IL-17, -23, -22) and JAK Inhibitors
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association studies |
| RA | Rheumatoid arthritis |
| HIES | Hyper immunoglobulin E syndrome |
References
- Christophers, E. Psoriasis—Epidemiology and clinical spectrum. Clin. Exp. Dermatol. 2001, 26, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, T.D.; Schupp, C.W.; Armstrong, A.W. Psoriasis prevalence among adults in the United States. J. Am. Acad. Dermatol. 2014, 70, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Troxel, A.B.; Lewis, J.D.; Kurd, S.K.; Shin, D.B.; Wang, X.; Margolis, D.J.; Strom, B.L. The risk of mortality in patients with psoriasis: Results from a population-based study. Arch. Dermatol. 2007, 143, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, J.; Grewal, S.; Langan, S.M.; Mehta, N.N.; Ogdie, A.; Van Voorhees, A.S.; Gelfand, J.M. Psoriasis and comorbid diseases: Epidemiology. J. Am. Acad. Dermatol. 2017, 76, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Sarkar, M.K.; Tsoi, L.C.; Gudjonsson, J.E. Psoriasis: A mixed autoimmune and autoinflammatory disease. Curr. Opin. Immunol. 2017, 49, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tiilikainen, A.; Lassus, A.; Karvonen, J.; Vartiainen, P.; Julin, M. Psoriasis and HLA-Cw6. Br. J. Dermatol. 1980, 102, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Stuart, P.E.; Nistor, I.; Hiremagalore, R.; Chia, N.V.C.; Jenisch, S.; Weichenthal, M.; Abecasis, G.R.; Lim, H.W.; Christophers, E.; et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am. J. Hum. Genet. 2006, 78, 827–851. [Google Scholar] [CrossRef] [PubMed]
- Henseler, T.; Christophers, E. Psoriasis of early and late onset: Characterization of two types of psoriasis vulgaris. J. Am. Acad. Dermatol. 1985, 13, 450–456. [Google Scholar] [CrossRef]
- Strange, A.; Capon, F.; Spencer, C.C.; Knight, J.; Weale, M.E.; Allen, M.H.; Barton, A.; Band, G.; Bellenguez, C.; Bergboer, J.G.M.; et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, J.E.; Johnston, A.; Sigmundsdottir, H.; Valdimarsson, H. Immunopathogenic mechanisms in psoriasis. Clin Exp. Immunol. 2004, 135, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.D.; Hagenaars, C.; Das, P.K.; Krieg, S.R.; Voorn, W.J.; Kapsenberg, M.L. Predominance of “memory” T cells (CD4+, CDw29+) over “naive” T cells (CD4+, CD45R+) in both normal and diseased human skin. Arch. Dermatol. Res. 1989, 281, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Smith, L.R.; Froning, K.J.; Schwabe, B.J.; Laxer, J.A.; Caralli, L.L.; Kurland, H.H.; Karasek, M.A.; Wilkinson, D.J.; Carlo, D.J. CD8+ T cells in psoriatic lesions preferentially use T-cell receptor V beta 3 and/or V beta 13.1 genes. Proc. Natl. Acad. Sci. USA 1994, 91, 9282–9286. [Google Scholar] [CrossRef] [PubMed]
- Res, P.C.M.; Piskin, G.; de Boer, O.J.; van der Loos, C.M.; Teeling, P.; Bos, J.D.; Teunissen, M.B.M. Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS ONE 2010, 5, e14108. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Ellinghaus, E.; Nair, R.P.; Stuart, P.E.; Esko, T.; Metspalu, A.; Debrus, S.; Raelson, J.V.; Tejasvi, T.; Belouchi, M.; et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am. J. Hum. Genet. 2012, 90, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Stuart, P.E.; Sarkar, M.K.; Voorhees, J.J.; Elder, J.T.; Johnston, A.; Gudjonsson, J.E. Cellular dissection of psoriasis for transcriptome analyses and the post-GWAS era. BMC Med. Genom. 2014, 7, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Wang, G.; Fan, J.Y.; Li, W.; Liu, Y.F. HLA DR B1*04, *07-restricted epitopes on Keratin 17 for autoreactive T cells in psoriasis. J. Dermatol. Sci. 2005, 38, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Valdimarsson, H.; Thorleifsdottir, R.H.; Sigurdardottir, S.L.; Gudjonsson, J.E.; Johnston, A. Psoriasis—As an autoimmune disease caused by molecular mimicry. Trends Immunol. 2009, 30, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wang, G. Keratin 17: A critical player in the pathogenesis of psoriasis. Med. Res. Rev. 2014, 34, 438–454. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, A.; Siewert, K.; Stohr, J.; Besgen, P.; Kim, S.M.; Rühl, G.; Nickel, J.; Vollmer, S.; Thomas, P.; Krebs, S.; et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J. Exp. Med. 2015, 212, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Jarrett, R.; Subramaniam, S.; Salimi, M.; Gutowska-Owsiak, D.; Chen, Y.L.; Hardman, C.; Xue, L.; Cerundolo, V.; Ogg, G. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J. Exp. Med. 2016, 213, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Conrad, C.; Dudli, C.; Kielhorn, E.; Nickoloff, B.J.; Nestle, F.O. Activation of dendritic antigen-presenting cells expressing common heat shock protein receptor CD91 during induction of psoriasis. Br. J. Dermatol. 2005, 152, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Zaba, L.C.; Fuentes-Duculan, J.; Eungdamrong, N.J.; Abello, M.V.; Novitskaya, I.; Pierson, K.C.; Gonzalez, J.; Krueger, J.G.; Lowes, M.A. Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J. Investig. Dermatol. 2009, 129, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Zaba, L.C.; Suarez-Farinas, M.; Fuentes-Duculan, J.; Nograles, K.E.; Guttman-Yassky, E.; Cardinale, I.; Lowes, M.A.; Krueger, J.G. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J. Allergy Clin. Immunol. 2009, 124, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Suarez-Farinas, M.; Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Hashimoto, M.; Yoshitomi, H.; Tanaka, S.; Nomura, T.; Yamaguchi, T.; Iwakura, Y.; Sakaguchi, N.; Sakaguchi, S. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J. Exp. Med. 2007, 204, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Sonderegger, I.; Rohn, T.A.; Kurrer, M.O.; Iezzi, G.; Zou, Y.; Kastelein, R.A.; Bachmann, F.M.; Kopf, M. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur. J. Immunol. 2006, 36, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.D.; Hulsebosch, H.J.; Krieg, S.R.; Bakker, P.M.; Cormane, R.H. Immunocompetent cells in psoriasis. In situ immunophenotyping by monoclonal antibodies. Arch. Dermatol. Res. 1983, 275, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Russell, C.B.; Martin, D.A.; Towne, J.E.; Krueger, J.G. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013, 34, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.; Wang, T.; Zheng, J.; et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Pantelyushin, S.; Croxford, A.L.; Kulig, P.; Becher, B. Dermal IL-17-producing gammadelta T cells establish long-lived memory in the skin. Eur. J. Immunol. 2015, 45, 3022–3033. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Valle, F.; Gray, E.E.; Cyster, J.G. Inflammation induces dermal Vgamma4+ gammadeltaT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proc. Natl. Acad. Sci. USA 2015, 112, 8046–8051. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, S.; Laukens, D.; Peeters, H.; Melis, L.; Olievier, K.; Boon, N.; Verbruggen, G.; Vandesompele, J.; Elewaut, D.; De Vos, M. Differential mucosal expression of Th17-related genes between the inflamed colon and ileum of patients with inflammatory bowel disease. BMC Immunol. 2010, 11, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brustle, A.; Brenner, D.; Knobbe, C.B.; Lang, P.A.; Virtanen, C.; Hershenfield, B.M.; Reardon, C.; Lacher, S.M.; Ruland, J.; Ohashi, P.S.; et al. The NF-κB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J. Clin. Investig. 2012, 122, 4698–4709. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Ma, H.L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Harper, E.G.; Guo, C.; Rizzo, H.; Lillis, J.V.; Kurtz, S.E.; Skorcheva, I.; Purdy, D.; Fitch, E.; Iordanov, M.; Blauvelt, A. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: Implications for psoriasis pathogenesis. J. Investig. Dermatol. 2009, 129, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Baliwag, J.; Barnes, D.H.; Johnston, A. Cytokines in psoriasis. Cytokine 2015, 73, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2006, 445, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Geiger, R.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 2009, 10, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Trifari, S.; Kaplan, C.D.; Tran, E.H.; Crellin, N.K.; Spits, H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat. Immunol. 2009, 10, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Canavese, M.; Altruda, F.; Ruzicka, T.; Schauber, J. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis—A possible target for novel therapies? J. Dermatol. Sci. 2010, 58, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Wegenka, U.M.; Buschmann, J.; Lutticken, C.; Heinreich, P.C.; Horn, F. Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol. Cell. Biol. 1993, 13, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Deenick, E.K.; Avery, D.T.; Chan, A.; Berglund, L.J.; Ives, M.L.; Moens, L.; Stoddard, J.L.; Bustamante, J.; Boisson-Dupuis, S.; Tsumura, M.; et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J. Exp. Med. 2013, 210, 2739–2753. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.K.; Andraski, A.B.; Spolski, R.; Li, P.; Kazemian, M.; Oh, J.; Samsel, L.; Swanson, P.A., II; McGavern, D.B.; Sampaio, E.P.; et al. Opposing roles of STAT1 and STAT3 in IL-21 function in CD4+ T cells. Proc. Natl. Acad. Sci. USA 2015, 112, 9394–9399. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, B.; Dang, E.; Jin, L.; Fan, X.; Wang, G. Impaired function of regulatory T cells in patients with psoriasis is mediated by phosphorylation of STAT3. J. Dermatol. Sci. 2016, 81, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, M.L.; Rascle, A.; Zurawski, S.; Menon, S.; de Waal Malefyt, R. Interleukin-22 activates STAT3 and induces IL-10 by colon epithelial cells. Int. Immunopharmacol. 2004, 4, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Perusina Lanfranca, M.; Lin, Y.; Fang, J.; Zou, W.; Frankel, T. Biological and pathological activities of interleukin-22. J. Mol. Med. 2016, 94, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Floss, D.M.; Schroder, J.; Franke, M.; Scheller, J. Insights into IL-23 biology: From structure to function. Cytokine Growth Factor Rev. 2015, 26, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed]
- Sa, S.M.; Valdez, P.A.; Wu, J.; Jung, K.; Zhong, F.; Hall, L.; Kasman, I.; Winer, J.; Modrusan, Z.; Danilenko, D.M.; et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J. Immunol. 2007, 178, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Camporeale, A.; Poli, V. IL-6, IL-17 and STAT3: A holy trinity in auto-immunity? Front. Biosci. 2012, 17, 2306–2326. [Google Scholar] [CrossRef]
- Camporeale, A.; Marino, F.; Papageorgiou, A.; Carai, P.; Fornero, S.; Fletcher, S.; Page, B.D.; Gunning, P.; Forni, M.; Chiarle, R.; et al. STAT3 activity is necessary and sufficient for the development of immune-mediated myocarditis in mice and promotes progression to dilated cardiomyopathy. EMBO Mol. Med. 2013, 5, 572–590. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Chan, K.S.; Carbajal, S.; Clifford, J.; Peavey, M.; Kiguchi, K.; Itami, S.; Nickoloff, B.J.; DiGiovanni, J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat. Med. 2004, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Haapaniemi, E.; Russell, M.A.; Caswell, R.; Allen, H.L.; De Franco, E.; McDonald, T.J.; Rajala, H.; Ramelius, A.; Barton, J.; et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat. Genet. 2014, 46, 812–814. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Vallania, F.; Schiavone, D.; Dewilde, S.; Pupo, E.; Garbay, S.; Calogero, R.; Pontoglio, M.; Provero, P.; Poli, V. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: The case of Stat3. Proc. Natl. Acad. Sci. USA 2009, 106, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol Chem 2002, 277, 8004–8011. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, F.; Lee, S.O.; Onate, S.A.; Gao, A.C. Stat3 enhances transactivation of steroid hormone receptors. Nucl. Recept. 2003, 1, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Boldogh, I.; Brasier, A.R. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 2005, 129, 1616–1632. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Ghoreschi, K.; Steward-Tharp, S.M.; Rodriguez-Canales, J.; Zhu, J.; Grainger, J.R.; Hirahara, K.; Sun, H.W.; Wei, L.; Vahedi, G.; et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011, 12, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Zugowski, C.; Lieder, F.; Muller, A.; Gasch, J.; Corvinus, F.M.; Moriggl, R.; Friedrich, K. STAT3 controls matrix metalloproteinase-1 expression in colon carcinoma cells by both direct and AP-1-mediated interaction with the MMP-1 promoter. Biol. Chem. 2011, 392, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, M.; Ogura, H.; Ueda, N.; Tsuruoka, M.; Kitabayashi, C.; Tsuji, F.; Aono, H.; Ishihara, K.; Huseby, E.; Betz, U.A.; et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 2007, 19, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L.; et al. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, Y.; Saito, M.; Tsuchiya, S.; Tsuge, I.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; Pasic, S.; Stojkovic, O.; et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007, 448, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Grosso, J.F.; Yen, H.R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Laurence, A.; Kanno, Y.; Pacher-Zavisin, M.; Zhu, B.M.; Tato, C.; Yoshimura, A.; Hennighausen, L.; O’Shea, J.J. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8137–8142. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Spolski, R.; Casas, E.; Zhu, W.; Levy, D.E.; Leonard, W.J. The molecular basis of IL-21-mediated proliferation. Blood 2007, 109, 4135–4142. [Google Scholar] [CrossRef] [PubMed]
- Akimzhanov, A.M.; Yang, X.O.; Dong, C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J. Biol. Chem. 2007, 282, 5969–5972. [Google Scholar] [CrossRef] [PubMed]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, J.; Mao, X.; Tang, Q.; Lu, H. IL-7 receptor blockade inhibits IL-17-producing gammadelta cells and suppresses melanoma development. Inflammation 2014, 37, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Gu, J.; Xiao, H.; Liang, S.; Yang, E.; Yang, R.; Huang, D.; Chen, C.; Wang, F.; Shen, L.; et al. Selective Destruction of Interleukin 23-Induced Expansion of a Major Antigen-Specific gammadelta T-Cell Subset in Patients With Tuberculosis. J. Infect. Dis. 2017, 215, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.L.; Yang, H.Y.; Wang, F.F.; Zhang, X.X.; Bai, Y.P. Interleukin-21 is associated with the severity of psoriasis vulgaris through promoting CD4+ T cells to differentiate into Th17 cells. Am. J. Transl. Res. 2016, 8, 3188–3196. [Google Scholar] [PubMed]
- Caruso, R.; Botti, E.; Sarra, M.; Esposito, M.; Stolfi, C.; Diluvio, L.; Giustizieri, M.L.; Pacciani, V.; Mazzotta, A.; Campione, E.; et al. Involvement of interleukin-21 in the epidermal hyperplasia of psoriasis. Nat. Med. 2009, 15, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Sestito, R.; Madonna, S.; Scarponi, C.; Cianfarani, F.; Failla, C.M.; Cavani, A.; Girolomoni, G.; Albanesi, C. STAT3-dependent effects of IL-22 in human keratinocytes are counterregulated by sirtuin 1 through a direct inhibition of STAT3 acetylation. FASEB J. 2011, 25, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Haugen, H.S.; Xu, W.; Witte, E.; Waggie, K.; Anderson, M.; Vom Baur, E.; Witte, K.; Warszawska, K.; Philipp, S.; et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-γ are not. J. Mol. Med. 2009, 87, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Witte, E.; Kokolakis, G.; Witte, K.; Philipp, S.; Doecke, W.D.; Babel, N.; Wittig, B.M.; Warszawska, K.; Kurek, A.; Erdmann-Keding, M.; et al. IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J. Investig. Dermatol. 2014, 134, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Komine, M.; Freedberg, I.M.; Blumenberg, M. Regulation of epidermal expression of keratin K17 in inflammatory skin diseases. J. Investig. Dermatol. 1996, 107, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dang, E.; Shi, X.; Jin, L.; Feng, Z.; Hu, L.; Wu, Y.; Wang, G. The Pro-Inflammatory Cytokine IL-22 Up-Regulates Keratin 17 Expression in Keratinocytes via STAT3 and ERK1/2. PLoS ONE 2012, 7, e40797–e40798. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, Y.; Cao, K.; Xu, Y.Y.; Gao, X.H.; Chen, H.D.; Geng, L. Shikonin inhibits IFN-gamma-induced K17 over-expression of HaCaT cells by interfering with STAT3 signaling. Int. J. Clin. Exp. Pathol. 2015, 8, 9202–9207. [Google Scholar] [PubMed]
- Shi, X.; Jin, L.; Dang, E.; Chang, T.; Feng, Z.; Liu, Y.; Wang, G. IL-17A Upregulates Keratin 17 Expression in Keratinocytes through STAT1- and STAT3-Dependent Mechanisms. J. Investig. Dermatol. 2011, 131, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, G. Keratin 17 as a therapeutic target for the treatment of psoriasis. J. Dermatol. Sci. 2012, 67, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, V.; Regis, G.; Tassone, B.; Valenti, C.; Avalle, L.; Saoncella, S.; Calautti, E.; Poli, V. Constitutive STAT3 activation in epidermal keratinocytes enhances cell clonogenicity and favours spontaneous immortalization by opposing differentiation and senescence checkpoints. Exp. Dermatol. 2015, 24, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bonnet, M.C.; Ulvmar, M.H.; Wolk, K.; Karagianni, N.; Witte, E.; Uthoff-Hachenberg, C.; Renauld, J.C.; Kollias, G.; Toftgard, R.; et al. Tumor Necrosis Factor Receptor Signaling in Keratinocytes Triggers Interleukin-24-Dependent Psoriasis-like Skin Inflammation in Mice. Immunity 2013, 39, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Goodman, W.A.; Levine, A.D.; Massari, J.V.; Sugiyama, H.; McCormick, T.S.; Cooper, K.D. IL-6 Signaling in Psoriasis Prevents Immune Suppression by Regulatory T Cells. J. Immunol. 2009, 183, 3170–3176. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Kanda, T.; Tarutani, M.; Iiyama, T.; Asao, N.; DiGiovanni, J.; Sano, S. Stat3 as a Therapeutic Target for the Treatment of Psoriasis: A Clinical Feasibility Study with STA-21, a Stat3 Inhibitor. J. Investig. Dermatol. 2011, 131, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kanda, T.; Takaishi, M.; Shiga, T.; Miyoshi, K.; Nakajima, H.; Kamijima, R.; Tarutani, M.; Benson, J.M.; Elloso, M.M.; et al. Distinct Roles of IL-23 and IL-17 in the Development of Psoriasis-Like Lesions in a Mouse Model. J. Immunol. 2011, 186, 4481–4489. [Google Scholar] [CrossRef] [PubMed]
- Jabbar-Lopez, Z.K.; Yiu, Z.Z.N.; Ward, V.; Exton, L.S.; Mohd Mustapa, M.F.; Samarasekera, E.; Burden, A.D.; Murphy, R.; Owen, C.M.; Parslew, R.; et al. Quantitative Evaluation of Biologic Therapy Options for Psoriasis: A Systematic Review and Network Meta-Analysis. J. Investig. Dermatol. 2017, 137, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Leonardi, C.; Papp, K.; Gottlieb, A.; Thaçi, D.; Schacht, A.; Ball, S.; Agada, N.; Mallbris, L. Comment on “Quantitative Evaluation of Biologic Therapy Options for Psoriasis: A Systematic Review and Network Meta-Analysis”. J. Investig. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.J.; Wang, T. Advances of the interleukin-21 signaling pathway in immunity and angiogenesis. Biomed. Rep. 2016, 5, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.; King, B.A. JAK inhibitors in dermatology: The promise of a new drug class. J. Am. Acad Dermatol. 2017, 76, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Wcislo-Dziadecka, D.; Zbiciak-Nylec, M.; Brzezinska-Wcislo, L.; Bebenek, K.; Kaźmierczak, A. Newer treatments of psoriasis regarding IL-23 inhibitors, phosphodiesterase 4 inhibitors, and Janus kinase inhibitors. Dermatol. Ther. 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Welsch, K.; Holstein, J.; Laurence, A.; Ghoreschi, K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur J. Immunol. 2017, 47, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Strand, V.; Kremer, J.M.; Gruben, D.; Krishnaswami, S.; Zwillich, S.H.; Wallenstein, G.V. Tofacitinib in Combination With Conventional Disease-Modifying Antirheumatic Drugs in Patients With Active Rheumatoid Arthritis: Patient-Reported Outcomes From a Phase III Randomized Controlled Trial. Arthritis Care Res. 2017, 69, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Wollenhaupt, J.; Silverfield, J.; Lee, E.B.; Curtis, J.R.; Wood, S.P.; Soma, K.; Nduaka, C.I.; Benda, B.; Gruben, D.; Nakamura, H.; et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J. Rheumatol. 2014, 41, 837–852. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K. Janus kinase inhibitors for rheumatoid arthritis. Curr. Opin. Chem. Biol. 2016, 32, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Di Lernia, V.; Bardazzi, F. Profile of tofacitinib citrate and its potential in the treatment of moderate-to-severe chronic plaque psoriasis. Drug Des. Dev. Ther. 2016, 10, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Colbert, R.A.; Ward, M.M. JAK Inhibitors Taking on Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1582–1584. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.; Rigby, W.; Azevedo, V.F.; Behrens, F.; Blanco, R.; Kaszuba, A.; Kudlacz, E.; Wang, C.; Menon, S.; Hendrikx, T.; et al. Tofacitinib for Psoriatic Arthritis in Patients with an Inadequate Response to TNF Inhibitors. N. Engl. J. Med. 2017, 377, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Hall, S.; FitzGerald, O.; van der Heijde, D.; Merola, J.; Avila-Zapata, F.; Cieślak, D.; Graham, D.; Wang, C.; Menon, S.; et al. Tofacitinib or Adalimumab versus Placebo for Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Bachelez, H.; van de Kerkhof, P.C.; Strohal, R.; Kubanov, A.; Valenzuela, F.; Lee, J.H.; Yakusevich, V.; Chimenti, S.; Papacharalambous, J.; Proulx, J.; et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: A phase 3 randomised non-inferiority trial. Lancet 2015, 386, 552–561. [Google Scholar] [CrossRef]
- Papp, K.A.; Menter, M.A.; Abe, M.; Elewski, B.; Feldman, S.R.; Gottlieb, A.B.; Langley, R.; Luger, T.; Thaci, D.; Buonanno, M.; et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: Results from two randomized, placebo-controlled, phase III trials. Br. J. Dermatol. 2015, 173, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.R.; Thaci, D.; Gooderham, M.; Augustin, M.; de la Cruz, C.; Mallbris, L.; Buonanno, M.; Tatulych, S.; Kaur, M.; Lan, S.; et al. Tofacitinib improves pruritus and health-related quality of life up to 52 weeks: Results from 2 randomized phase III trials in patients with moderate to severe plaque psoriasis. J. Am. Acad. Dermatol. 2016, 75, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Stander, S.; Luger, T.; Cappelleri, J.C.; Bushmakin, A.G.; Mamolo, C.; Zielinski, M.A.; Tallman, A.M.; Yosipovitch, G. Validation of the Itch Severity Item as a Measurement Tool for Pruritus in Patients with Psoriasis: Results from a Phase 3 Tofacitinib Program. Acta Derm. Venereol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Punwani, N.; Scherle, P.; Flores, R.; Shi, J.; Liang, J.; Yeleswaram, S.; Levy, R.; Williams, W.; Gottlieb, A. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J. Am. Acad. Dermatol. 2012, 67, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Alves de Medeiros, A.K.; Speeckaert, R.; Desmet, E.; Van Gele, M.; De Schepper, S.; Lambert, J. JAK3 as an Emerging Target for Topical Treatment of Inflammatory Skin Diseases. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cell Type | Cytokine | Effect | Refs. |
|---|---|---|---|
| Th17 | IL-6 | differentiation, induction of IL-23 receptor | [30,31] |
| IL-23 | amplification and maintainance | [78] | |
| IL-21 | differentiation | [79] | |
| IL-22 | cross-talk lymphocytes-epithelial cells | [46] | |
| IL6, IL-23 | activates IL-17A and IL-17F, RORγT, RORα, BATF, IRF4, AHR, IL-6Rα and C-MAF | [81] | |
| Keratinocyte | IL-21 | proliferation and epidermal hyperplasia | [85] |
| IL-22 | proliferation, reduced differentiation and acanthosis | [46,86,87] | |
| IL-19 | amplification of IL-23/IL-17 axis, and induction of pro-inflammatory mediators | [59,88] | |
| IL-22, IL-17 | KRT17 induction | [46,89,90,91,92,93] | |
| γδ T cells | IL-7 | expansion | [82] |
| IL-23 | expansion | [83] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calautti, E.; Avalle, L.; Poli, V. Psoriasis: A STAT3-Centric View. Int. J. Mol. Sci. 2018, 19, 171. https://doi.org/10.3390/ijms19010171
Calautti E, Avalle L, Poli V. Psoriasis: A STAT3-Centric View. International Journal of Molecular Sciences. 2018; 19(1):171. https://doi.org/10.3390/ijms19010171
Chicago/Turabian StyleCalautti, Enzo, Lidia Avalle, and Valeria Poli. 2018. "Psoriasis: A STAT3-Centric View" International Journal of Molecular Sciences 19, no. 1: 171. https://doi.org/10.3390/ijms19010171
APA StyleCalautti, E., Avalle, L., & Poli, V. (2018). Psoriasis: A STAT3-Centric View. International Journal of Molecular Sciences, 19(1), 171. https://doi.org/10.3390/ijms19010171