Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients

 and
and 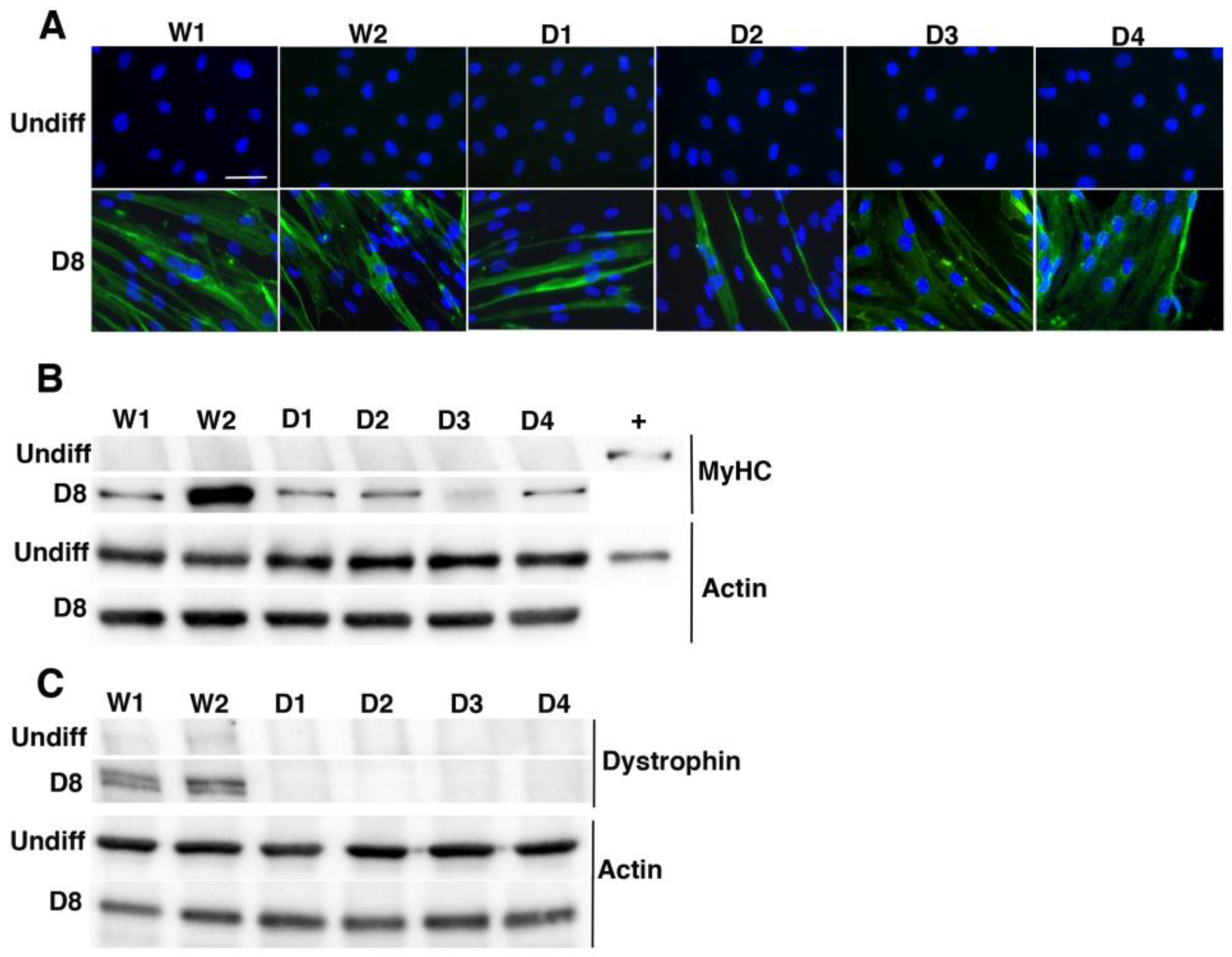{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Characterization of Immortalized Myoblasts Derived from Healthy Donors or Duchenne Muscular Dystrophy Patients
2.2. Protein Aggregation Is Increased in DMD Myoblasts
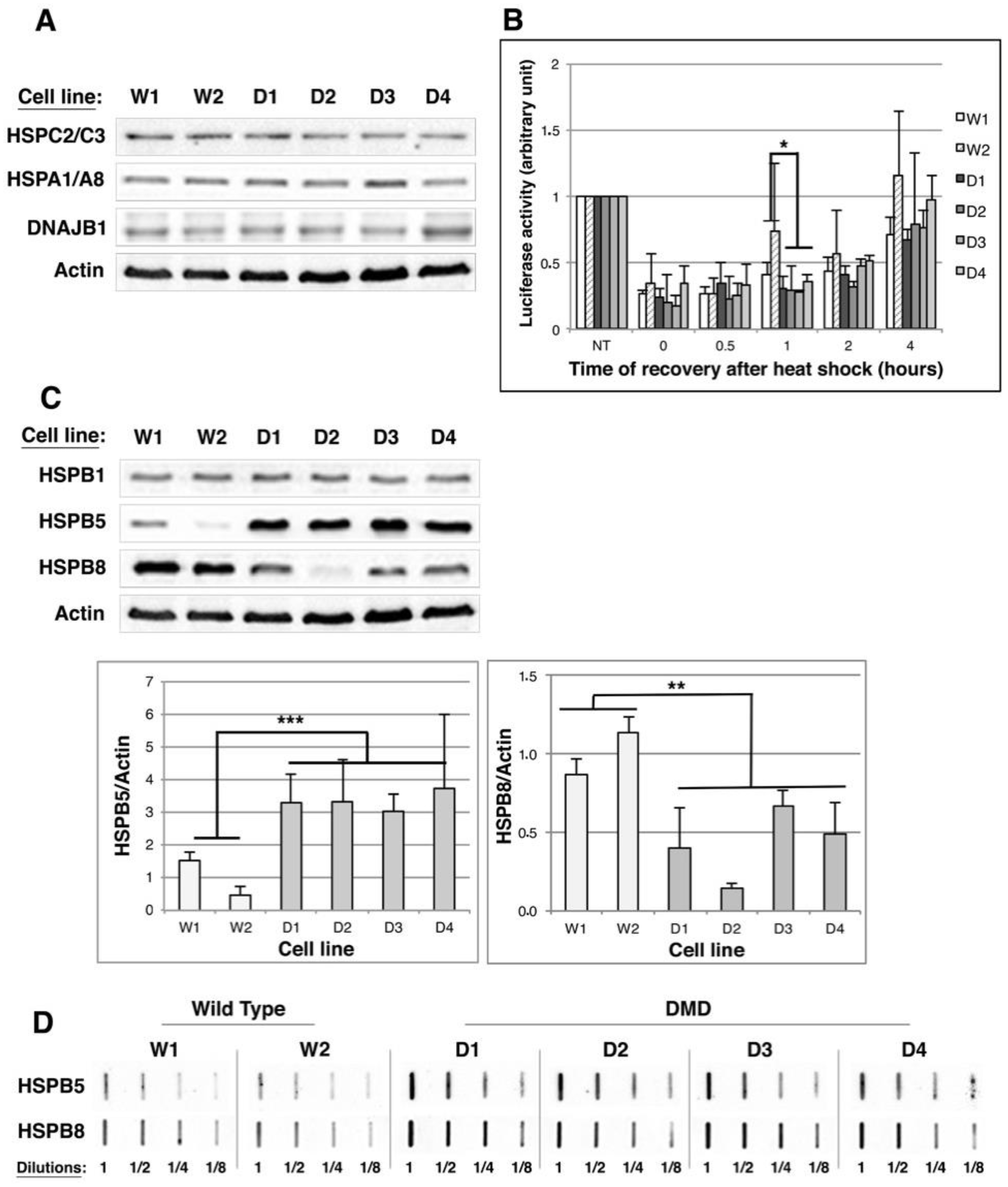
2.3. HSPB5 and HSPB8 Levels Are Modulated in DMD Myoblasts
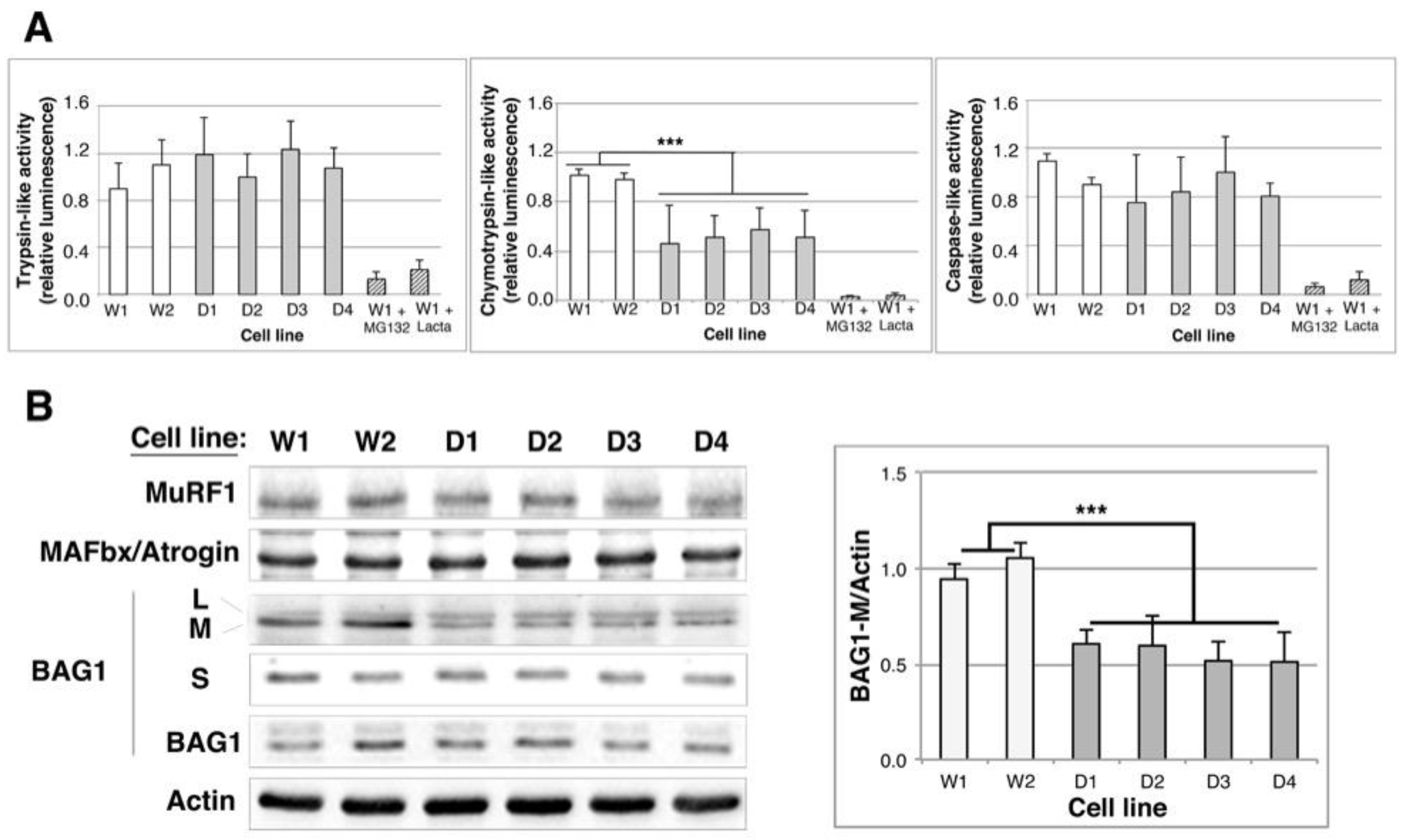
2.4. Proteasome Activity Is Decreased in DMD Myoblasts
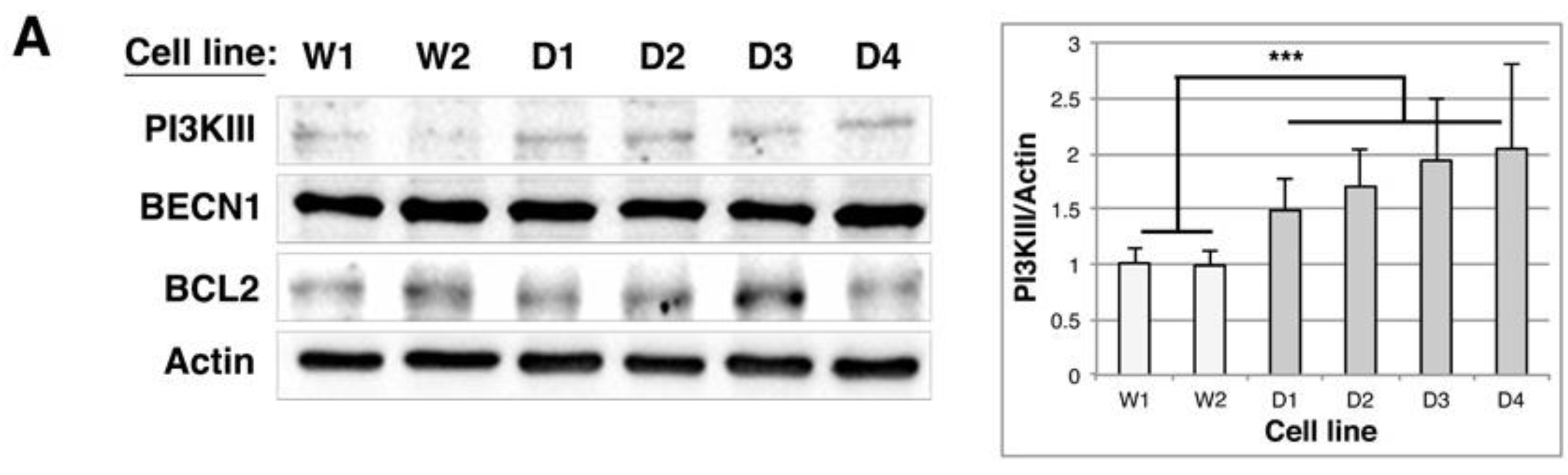
2.5. BAG1/BAG3 Ratio Is Inverted, BAG3/HSPB8 Complexes Are Increased and Autophagy Is Up-Regulated in DMD Myoblasts
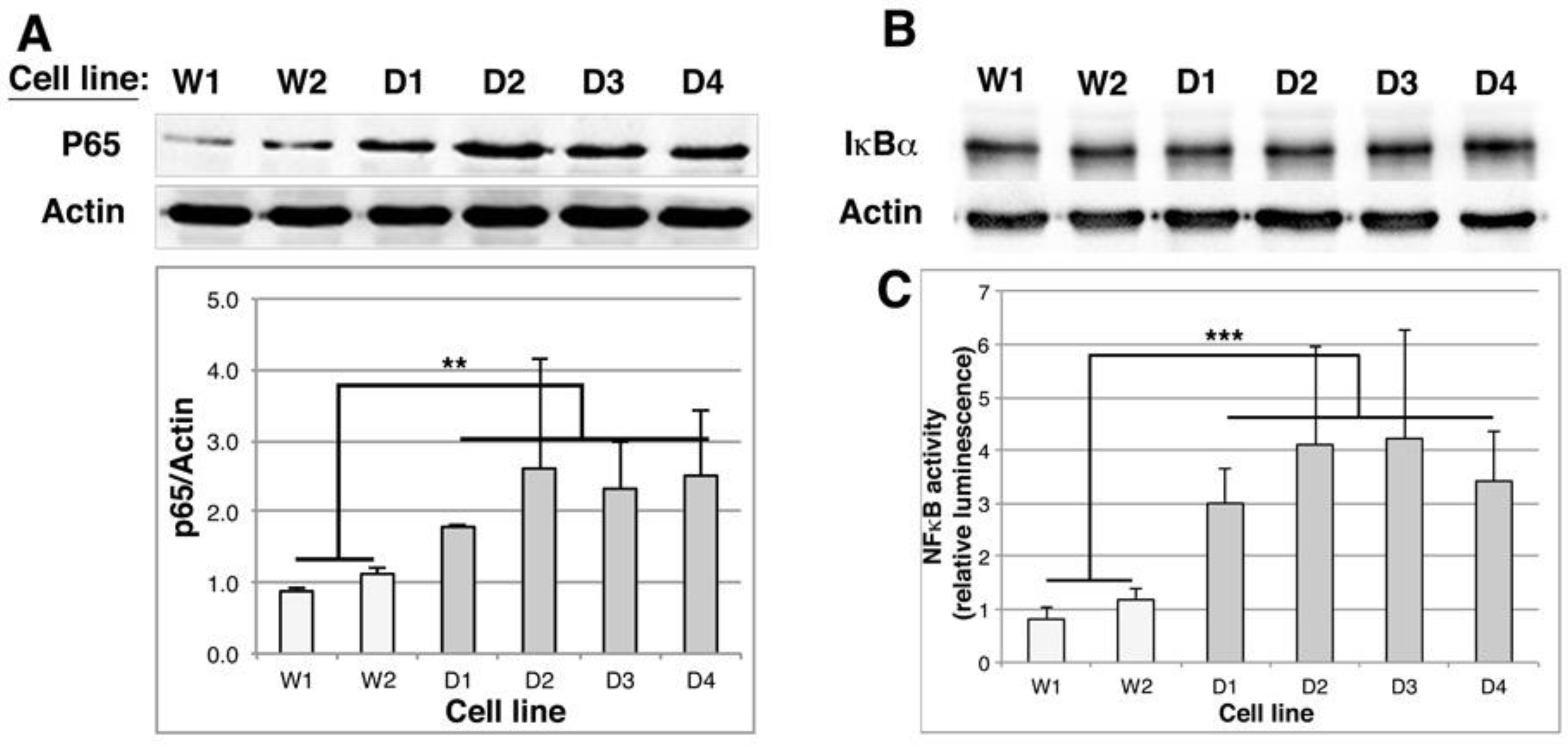
2.6. NFκB Activity Is Stimulated in DMD Myoblasts
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Lines
4.2. Reagents and Plasmids
4.3. Transfection
4.4. Gel Electrophoresis and Western-Blot
4.5. Proteasome Assay
4.6. Luciferase Assay
4.7. Luciferase Refolding Assay
4.8. Filter Trap Assay
4.9. Fluorescence Microscopy Analysis
4.10. Proximity Ligation Assay (PLA)
4.11. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DMD | Duchenne Muscular Dystrophy |
| HSP | Het Shock Protein |
| MyHC | Myosin Heavy Chains |
| PQC | Protein Quality Control |
| Ub | Ubiquitin |
| UPS | Ubiquitin Proteasome System |
References
- Herczenik, E.; Gebbink, M.F. Molecular and cellular aspects of protein misfolding and disease. FASEB J. 2008, 22, 2115–2133. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell. 2010, 40, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 2011, 3, a004374. [Google Scholar] [CrossRef] [PubMed]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb. Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef] [PubMed]
- Suss, O.; Reichmann, D. Protein plasticity underlines activation and function of atp-independent chaperones. Front. Mol. Biosci. 2015, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Kettern, N.; Dreiseidler, M.; Tawo, R.; Hohfeld, J. Chaperone-assisted degradation: Multiple paths to destruction. Biol. Chem. 2010, 391, 4814–4889. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.P.; Tanaka, K.; Goldberg, A.L.; Welch, W.J. Identity of the 19s ‘prosome’ particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature 1988, 331, 1921–1994. [Google Scholar] [CrossRef] [PubMed]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26s proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Dick, T.P.; Nussbaum, A.K.; Deeg, M.; Heinemeyer, W.; Groll, M.; Schirle, M.; Keilholz, W.; Stevanovic, S.; Wolf, D.H.; Huber, R.; et al. Contribution of proteasomal beta-subunits to the cleavage of peptide substrates analyzed with yeast mutants. J. Biol. Chem. 1998, 273, 25637–25646. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R., 3rd; Koonin, E.V.; Deshaies, R.J. Role of rpn11 metalloprotease in deubiquitination and degradation by the 26s proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Joachim, J.; Tooze, S.A. Autophagosome formation—The role of ulk1 and beclin1-pi3kc3 complexes in setting the stage. Semin. Cancer Biol. 2013, 23, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.I.; Dooley, H.C.; Tooze, S.A. Wipi2b and atg16l1: Setting the stage for autophagosome formation. Biochem. Soc. Trans. 2014, 42, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Muller, A.J.; Zuleger, T.; Takacs, Z.; Franz-Wachtel, M.; Thost, A.K.; Brigger, D.; Tschan, M.P.; Frickey, T.; Robenek, H.; et al. Wipi3 and wipi4 beta-propellers are scaffolds for lkb1-ampk-tsc signalling circuits in the control of autophagy. Nat. Commun. 2017, 8, 15637. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Nivon, M.; Fort, L.; Muller, P.; Richet, E.; Simon, S.; Guey, B.; Fournier, M.; Arrigo, A.P.; Hetz, C.; Atkin, J.D.; et al. Nfkappab is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation. Mol. Biol. Cell 2016, 27, 1712–1727. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Martens, S. Mechanisms of selective autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Sandri, M.; Coletto, L.; Grumati, P.; Bonaldo, P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the duchenne muscular dystrophy (dmd) cdna and preliminary genomic organization of the dmd gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Davies, K.E.; Smith, T.J.; Bundey, S.; Read, A.P.; Flint, T.; Bell, M.; Speer, A. Mild and severe muscular dystrophy associated with deletions in xp21 of the human x chromosome. J. Med. Genet. 1988, 25, 91–93. [Google Scholar] [CrossRef]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; Del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Bordoni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Perrotta, C.; Pellegrino, P.; Clementi, E.; Cervia, D. Skeletal muscle homeostasis in duchenne muscular dystrophy: Modulating autophagy as a promising therapeutic strategy. Front. Aging Neurosci. 2014, 6, 188. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Motohashi, Y.; Miyatake, S.; Komaki, H.; Takeda, S.; Aoki, Y. Recent advances in innovative therapeutic approaches for duchenne muscular dystrophy: From discovery to clinical trials. Am. J. Trans. Res. 2016, 8, 2471–2489. [Google Scholar]
- Carberry, S.; Brinkmeier, H.; Zhang, Y.; Winkler, C.K.; Ohlendieck, K. Comparative proteomic profiling of soleus, extensor digitorum longus, flexor digitorum brevis and interosseus muscles from the mdx mouse model of duchenne muscular dystrophy. Int. J. Mol. Med. 2013, 32, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Comparative proteomic analysis of the contractile-protein-depleted fraction from normal versus dystrophic skeletal muscle. Anal. Biochem. 2014, 446, 108–115. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Morisi, F.; Cheli, S.; Pambianco, S.; Cappello, V.; Vezzoli, M.; Rovere-Querini, P.; Moggio, M.; Ripolone, M.; Francolini, M.; et al. Autophagy as a new therapeutic target in duchenne muscular dystrophy. Cell Death Dis. 2012, 3, e418. [Google Scholar] [CrossRef] [PubMed]
- Brouilly, N.; Lecroisey, C.; Martin, E.; Pierson, L.; Mariol, M.C.; Qadota, H.; Labouesse, M.; Streichenberger, N.; Mounier, N.; Gieseler, K. Ultra-structural time-course study in the C. elegans model for duchenne muscular dystrophy highlights a crucial role for sarcomere-anchoring structures and sarcolemma integrity in the earliest steps of the muscle degeneration process. Hum. Mol. Genet. 2015, 24, 6428–6445. [Google Scholar] [CrossRef] [PubMed]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.; St Guily, J.L.; et al. Immortalized pathological human myoblasts: Towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle 2011, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Meer, D.P.; Eddinger, T.J. Heterogeneity of smooth muscle myosin heavy chain expression at the single cell level. Am. J. Physiol. 1996, 270, C1819–C1824. [Google Scholar] [CrossRef] [PubMed]
- Korfage, J.A.; Kwee, K.E.; Everts, V.; Langenbach, G.E. Myosin heavy chain expression can vary over the length of jaw and leg muscles. Cells Tissues Organs 2016, 201, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. Nbr1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Nicholl, I.D.; Quinlan, R.A. Chaperone activity of alpha-crystallins modulates intermediate filament assembly. EMBO J. 1994, 13, 945–953. [Google Scholar] [PubMed]
- Singh, B.N.; Rao, K.S.; Ramakrishna, T.; Rangaraj, N.; Rao Ch, M. Association of alphab-crystallin, a small heat shock protein, with actin: Role in modulating actin filament dynamics in vivo. J. Mol. Biol. 2007, 366, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Ohto, E.; Katayama, E.; Atomi, Y. Alphab-crystallin-coated map microtubule resists nocodazole and calcium-induced disassembly. J. Cell. Sci. 2004, 117, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by bag3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. Bag3 induces the sequestration of proteasomal clients into cytoplasmic puncta: Implications for a proteasome-to-autophagy switch. Autophagy 2014, 10, 1603–1621. [Google Scholar] [CrossRef] [PubMed]
- Nivon, M.; Abou-Samra, M.; Richet, E.; Guyot, B.; Arrigo, A.P.; Kretz-Remy, C. Nf-kappab regulates protein quality control after heat stress through modulation of the bag3-hspb8 complex. J. Cell Sci. 2012, 125, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, A.; Gehlert, S.; Leciejewski, B.; Schiffer, T.; Bloch, W.; Hohfeld, J. Induction and adaptation of chaperone-assisted selective autophagy casa in response to resistance exercise in human skeletal muscle. Autophagy 2015, 11, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Seguin, S.J.; Lambert, H.; Landry, J. Hspb8 chaperone activity toward poly(q)-containing proteins depends on its association with bag3, a stimulator of macroautophagy. J. Biol. Chem. 2008, 283, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Abu-Baker, A.; Messaed, C.; Laganiere, J.; Gaspar, C.; Brais, B.; Rouleau, G.A. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2003, 12, 2609–2623. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Bonaldo, P. Mitochondrial dysfunction and defective autophagy in the pathogenesis of collagen VI muscular dystrophies. Cold Spring Harb. Perspect. Biol. 2013, 5, a011387. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, T.; Fujimoto, S.; Ito, T.; Horinouchi, H.; Ueyama, H.; Tsuda, T. Proteasome expression in the skeletal muscles of patients with muscular dystrophy. Acta Neuropathol. 2000, 100, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Tashiro, Y.; Suzuki, N.; Warita, H.; Kato, M.; Tateyama, M.; Ando, R.; Izumi, R.; Yamazaki, M.; Abe, M.; et al. Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 2014, 127, 5204–5217. [Google Scholar] [CrossRef] [PubMed]
- Carmignac, V.; Svensson, M.; Korner, Z.; Elowsson, L.; Matsumura, C.; Gawlik, K.I.; Allamand, V.; Durbeej, M. Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of mdc1a. Hum. Mol. Genet. 2011, 20, 4891–4902. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, K.; Grisoni, K.; Segalat, L. Genetic suppression of phenotypes arising from mutations in dystrophin-related genes in caenorhabditis elegans. Curr. Biol. 2000, 10, 1092–1097. [Google Scholar] [CrossRef]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Takeda, S. Mammalian models of duchenne muscular dystrophy: Pathological characteristics and therapeutic applications. J. Biomed. Biotechnol. 2011, 2011, 184393. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, F.; Favaloro, A.; Fulle, S.; Magaudda, L.; Puglielli, C.; Di Mauro, D. Culture of human skeletal muscle myoblasts: Timing appearance and localization of dystrophin-glycoprotein complex and vinculin-talin-integrin complex. Cells Tissues Organs 2006, 183, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.F.; Bonilla, E.; Martucci, G.; Moraes, C.T.; Hays, A.P.; Dimauro, S. Immunocytochemical study of dystrophin in muscle cultures from patients with duchenne muscular dystrophy and unaffected control patients. Am. J. Pathol. 1988, 132, 410–416. [Google Scholar] [PubMed]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.M.; Lee, A.; Ervasti, J.M. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc. Natl. Acad. Sci. USA 2010, 107, 9632–9637. [Google Scholar] [CrossRef] [PubMed]
- Paepe, B.D.; Creus, K.K.; Weis, J.; Bleecker, J.L. Heat shock protein families 70 and 90 in duchenne muscular dystrophy and inflammatory myopathy: Balancing muscle protection and destruction. Neuromuscul. Disord. 2012, 22, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeier, H.; Ohlendieck, K. Chaperoning heat shock proteins: Proteomic analysis and relevance for normal and dystrophin-deficient muscle. Proteom. Clin. Appl. 2014, 8, 875–895. [Google Scholar] [CrossRef] [PubMed]
- Golenhofen, N.; Perng, M.D.; Quinlan, R.A.; Drenckhahn, D. Comparison of the small heat shock proteins alphab-crystallin, mkbp, hsp25, hsp20, and cvhsp in heart and skeletal muscle. Histochem. Cell Biol. 2004, 122, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.L.; Der Perng, M.; Prescott, A.R.; Jansen, K.A.; Koenderink, G.H.; Quinlan, R.A. The specificity of the interaction between alphab-crystallin and desmin filaments and its impact on filament aggregation and cell viability. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prevost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tome, F.; Dupret, J.M.; et al. A missense mutation in the alphab-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Capponi, S.; Geroldi, A.; Fossa, P.; Grandis, M.; Ciotti, P.; Gulli, R.; Schenone, A.; Mandich, P.; Bellone, E. Hspb1 and hspb8 in inherited neuropathies: Study of an italian cohort of dhmn and cmt2 patients. J. Peripher. Nerv. Syst. 2011, 16, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Nakhro, K.; Park, J.M.; Kim, Y.J.; Yoon, B.R.; Yoo, J.H.; Koo, H.; Choi, B.O.; Chung, K.W. A novel lys141thr mutation in small heat shock protein 22 (hspb8) gene in charcot-marie-tooth disease type 2l. Neuromuscul. Disord. 2013, 23, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, P.A.; Coyne, E.S.; Wing, S.S. The ubiquitin proteasome system in atrophying skeletal muscle: Roles and regulation. Am. J. Physiol. Cell Physiol. 2016, 311, C392–C403. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Assereto, S.; Piccirillo, R.; Baratto, S.; Scudieri, P.; Fiorillo, C.; Massacesi, M.; Traverso, M.; Galietta, L.J.; Bruno, C.; Minetti, C.; et al. The ubiquitin ligase tripartite-motif-protein 32 is induced in duchenne muscular dystrophy. Lab. Investig. 2016, 96, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Sotgia, F.; Capozza, F.; Gazzerro, E.; Minetti, C.; Lisanti, M.P. Localized treatment with a novel fda-approved proteasome inhibitor blocks the degradation of dystrophin and dystrophin-associated proteins in mdx mice. Cell Cycle 2007, 6, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.N.; Graber, T.G.; Bratten, W.M.; Ferrington, D.A.; Thompson, L.V. Immunoproteasome in animal models of duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2014, 35, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Luders, J.; Demand, J.; Papp, O.; Hohfeld, J. Distinct isoforms of the cofactor bag-1 differentially affect hsc70 chaperone function. J. Biol. Chem. 2000, 275, 14817–14823. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Demand, J.; Esser, C.; Emmerich, N.; Schild, H.; Hohfeld, J. Ubiquitylation of bag-1 suggests a novel regulatory mechanism during the sorting of chaperone substrates to the proteasome. J. Biol. Chem. 2002, 277, 45920–45927. [Google Scholar] [CrossRef] [PubMed]
- Demand, J.; Alberti, S.; Patterson, C.; Hohfeld, J. Cooperation of a ubiquitin domain protein and an e3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001, 11, 1569–1577. [Google Scholar] [CrossRef]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Pegoraro, E.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in bag3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Polanco, M.J.; Cristofani, R.; Cicardi, M.E.; Meroni, M.; Galbiati, M.; Piccolella, M.; Messi, E.; Giorgetti, E.; Lieberman, A.P.; et al. Aberrant autophagic response in the muscle of a knock-in mouse model of spinal and bulbar muscular atrophy. Sci. Rep. 2015, 5, 15174. [Google Scholar] [CrossRef] [PubMed]
- Bibee, K.P.; Cheng, Y.J.; Ching, J.K.; Marsh, J.N.; Li, A.J.; Keeling, R.M.; Connolly, A.M.; Golumbek, P.T.; Myerson, J.W.; Hu, G.; et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASEB J. 2014, 28, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Spitali, P.; Grumati, P.; Hiller, M.; Chrisam, M.; Aartsma-Rus, A.; Bonaldo, P. Autophagy is impaired in the tibialis anterior of dystrophin null mice. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, H.R.; Kelly, E.M.; Quindry, J.C.; Sheffield, J.B.; Hudson, M.B.; Selsby, J.T. Autophagic dysfunction and autophagosome escape in the mdx mus musculus model of duchenne muscular dystrophy. Acta Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kretz-Remy, C.; Bates, E.E.; Arrigo, A.P. Amino acid analogs activate nf-kappab through redox-dependent ikappab-alpha degradation by the proteasome without apparent ikappab-alpha phosphorylation. Consequence on hiv-1 long terminal repeat activation. J. Biol. Chem. 1998, 273, 3180–3191. [Google Scholar] [CrossRef] [PubMed]
- Monici, M.C.; Aguennouz, M.; Mazzeo, A.; Messina, C.; Vita, G. Activation of nuclear factor-kappab in inflammatory myopathies and duchenne muscular dystrophy. Neurology 2003, 60, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of tgfbeta in duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.; Carathers, M.; Li, Z.W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of ikk/nf-kappab signaling in macrophages and myofibers promotes muscle degeneration in duchenne muscular dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Kretz-Remy, C.; Munsch, B.; Arrigo, A.P. Nfkappa b-dependent transcriptional activation during heat shock recovery. Thermolability of the nf-kappab.Ikappa b complex. J. Biol. Chem. 2001, 276, 43723–43733. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Rojas, M.; Amair-Pinedo, F.; Tato, I.; Bartrons, R.; Ventura, F.; Rosa, J.L. Tris-acetate polyacrylamide gradient gels for the simultaneous electrophoretic analysis of proteins of very high and low molecular mass. Methods Mol. Biol. 2012, 869, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Nivon, M.; Richet, E.; Codogno, P.; Arrigo, A.P.; Kretz-Remy, C. Autophagy activation by nfkappab is essential for cell survival after heat shock. Autophagy 2009, 5, 766–783. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wattin, M.; Gaweda, L.; Muller, P.; Baritaud, M.; Scholtes, C.; Lozano, C.; Gieseler, K.; Kretz-Remy, C. Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients. Int. J. Mol. Sci. 2018, 19, 178. https://doi.org/10.3390/ijms19010178
Wattin M, Gaweda L, Muller P, Baritaud M, Scholtes C, Lozano C, Gieseler K, Kretz-Remy C. Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients. International Journal of Molecular Sciences. 2018; 19(1):178. https://doi.org/10.3390/ijms19010178
Chicago/Turabian StyleWattin, Marion, Loïc Gaweda, Pascale Muller, Mathieu Baritaud, Charlotte Scholtes, Chloé Lozano, Kathrin Gieseler, and Carole Kretz-Remy. 2018. "Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients" International Journal of Molecular Sciences 19, no. 1: 178. https://doi.org/10.3390/ijms19010178
APA StyleWattin, M., Gaweda, L., Muller, P., Baritaud, M., Scholtes, C., Lozano, C., Gieseler, K., & Kretz-Remy, C. (2018). Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients. International Journal of Molecular Sciences, 19(1), 178. https://doi.org/10.3390/ijms19010178