Aryl Hydrocarbon Receptor Antagonists Mitigate the Effects of Dioxin on Critical Cellular Functions in Differentiating Human Osteoblast-Like Cells

Abstract
:1. Introduction
2. Results
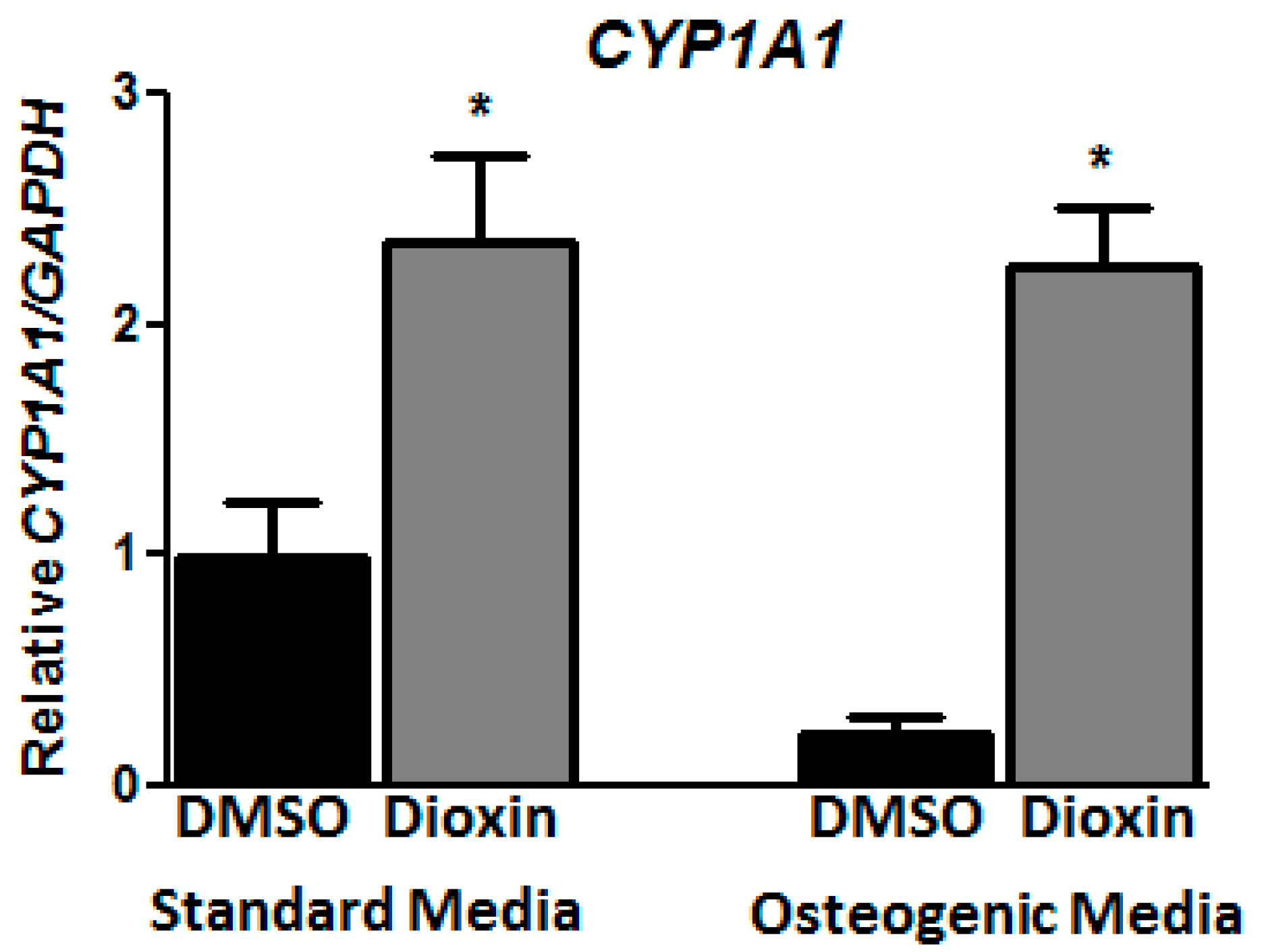
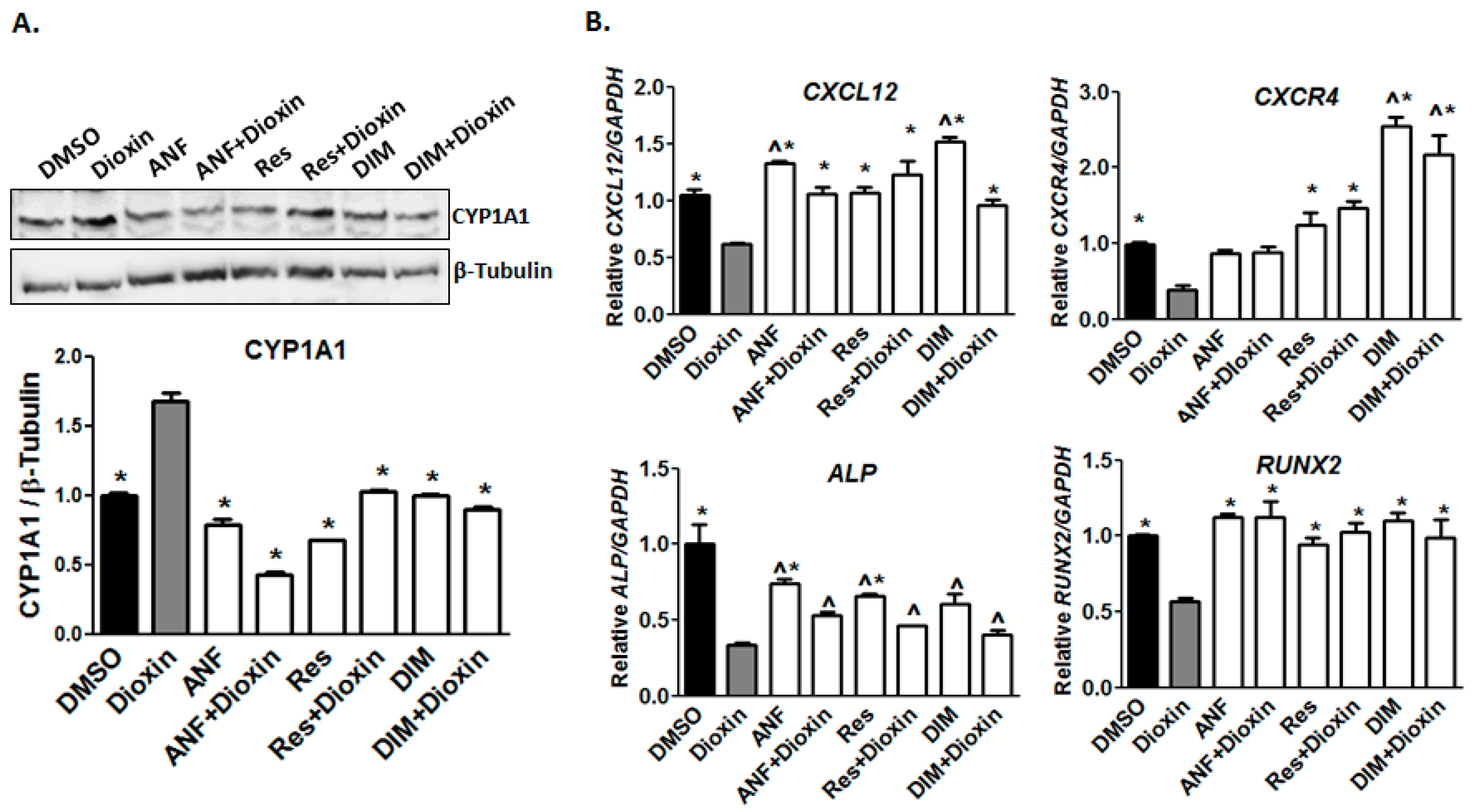
2.1. Dioxin Exposure and AhR Activation
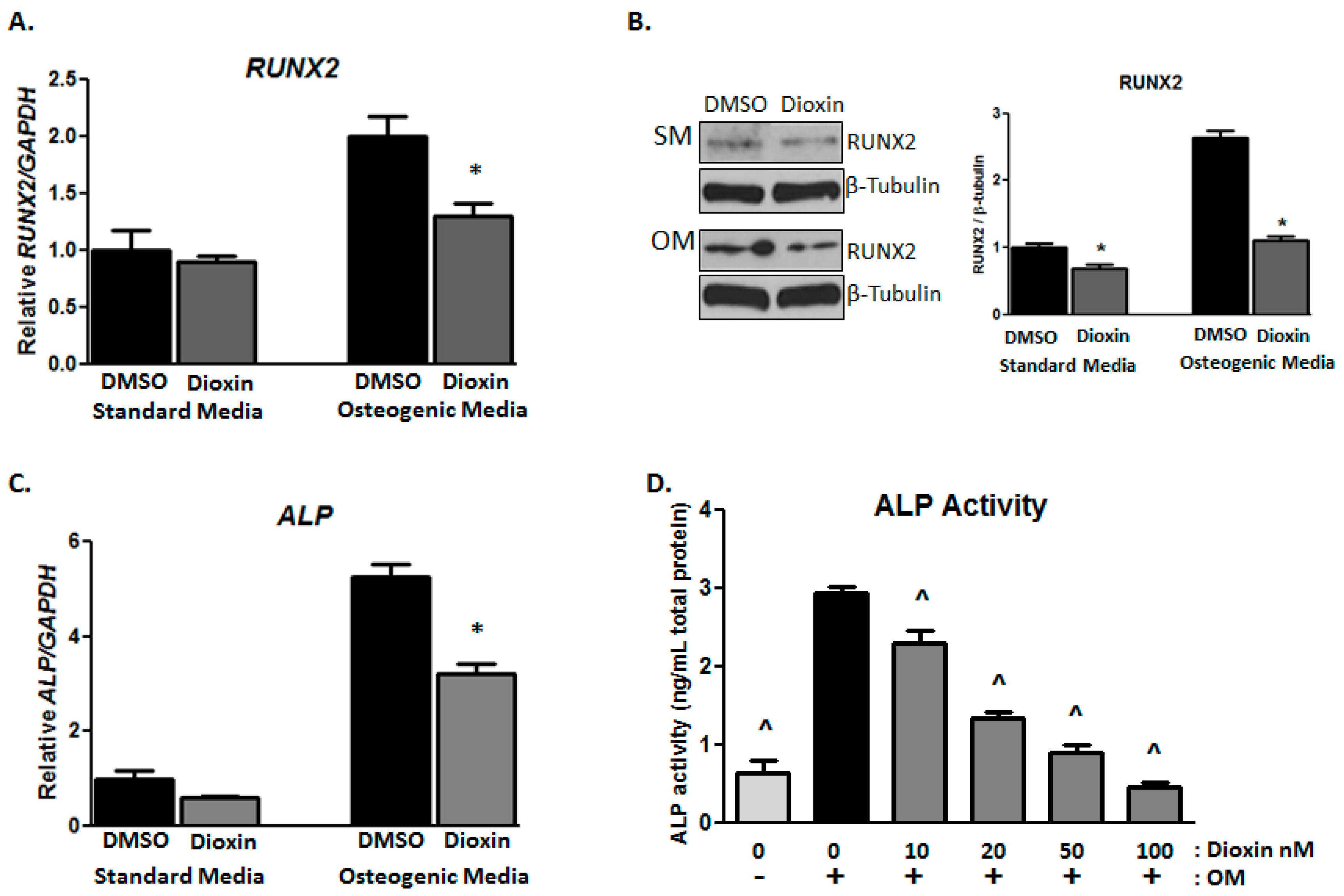
2.2. Early Markers of Osteogenic Differentiation
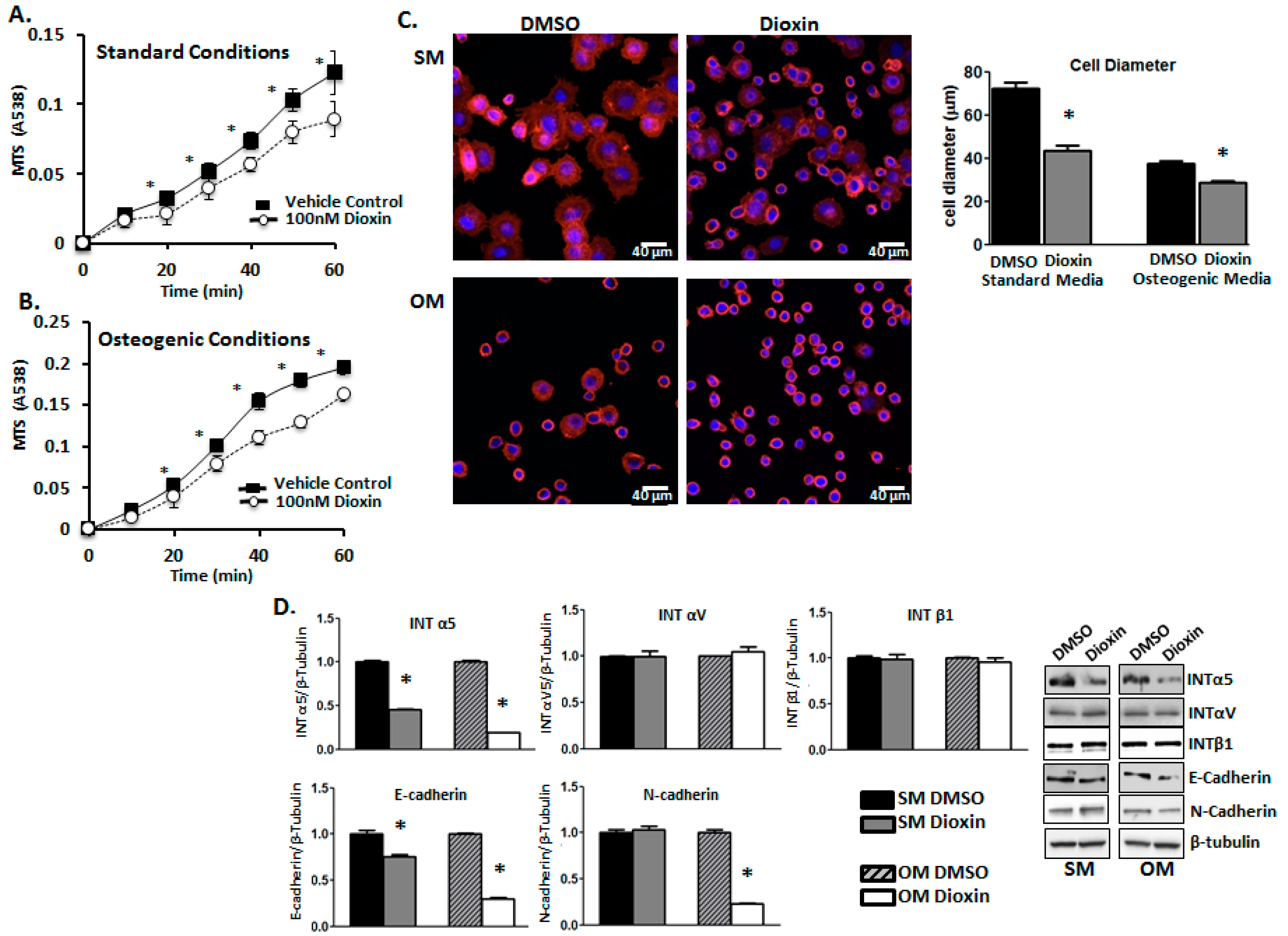
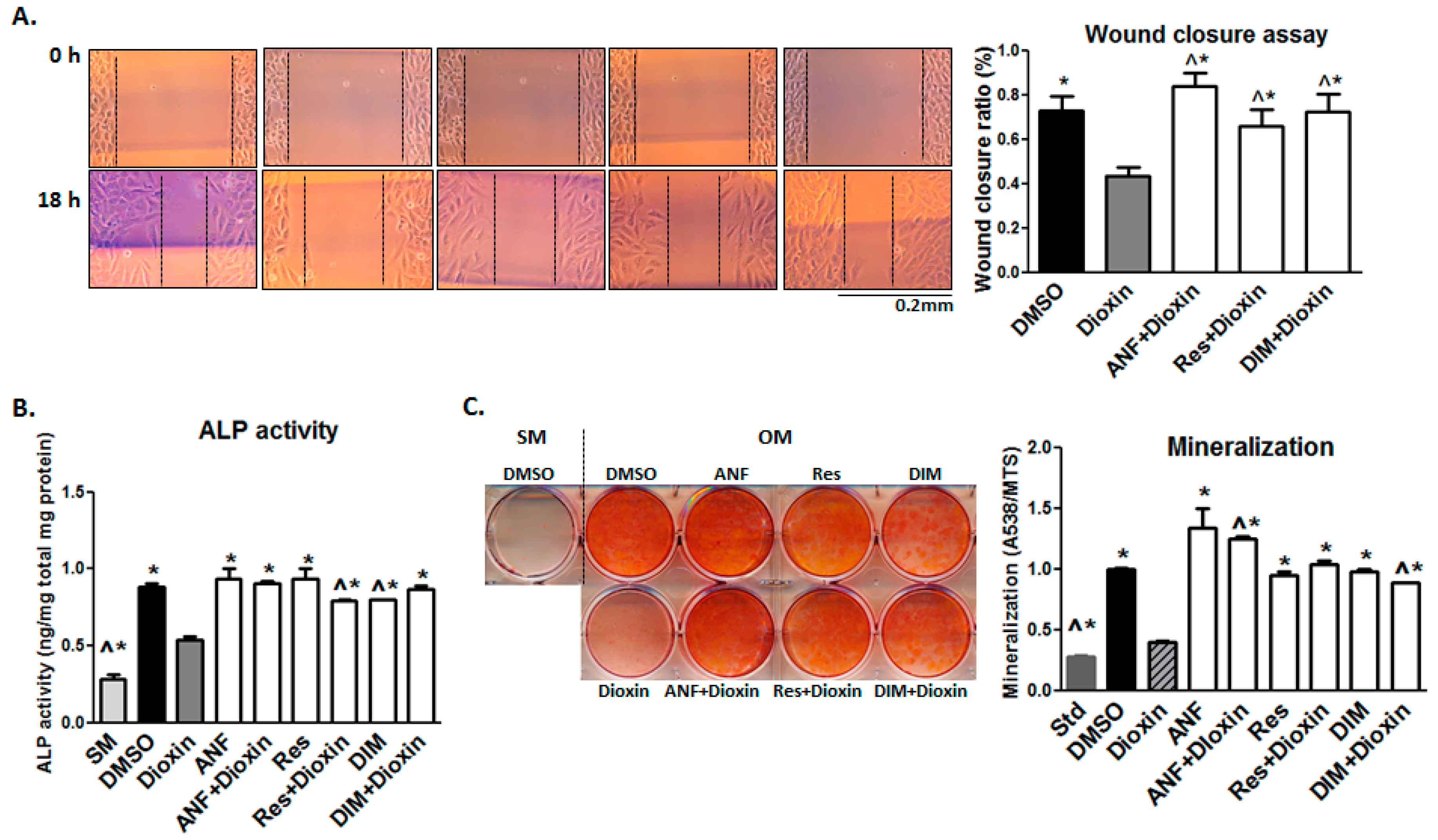
2.3. Cell Adhesion
2.4. Cell Migration
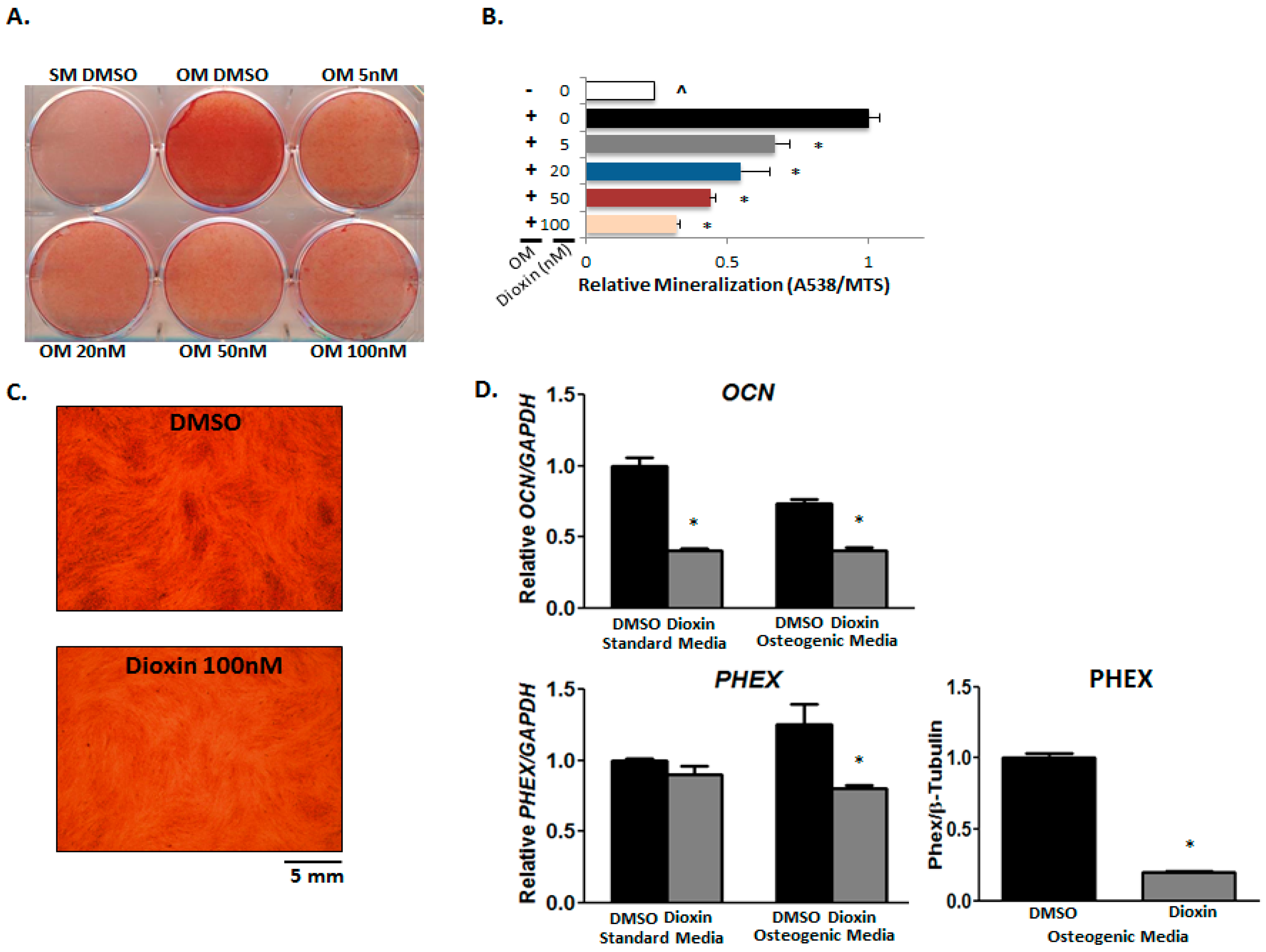
2.5. Matrix Mineralization
2.6. AhR Antagonist Studies
3. Discussion
4. Materials and Methods
4.1. MG-63 Cell Culture
4.2. Alkaline Phosphatase Activity Assays
4.3. Cell Adhesion Assays
4.4. Actin Filament Re-Polymerization Assay
4.5. Cell Migration Assays
4.6. Mineralization Assays
4.7. Western Blotting
4.8. RNA Isolation and Gene Expression Analysis
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Middlekauff, H.R.; Park, J.; Moheimani, R.S. Adverse effects of cigarette and noncigarette smoke exposure on the autonomic nervous system: Mechanisms and implications for cardiovascular risk. J. Am. Coll. Cardiol. 2014, 64, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Sasco, A.J.; Secretan, M.B.; Straif, K. Tobacco smoking and cancer: A brief review of recent epidemiological evidence. Lung Cancer 2004, 45 (Suppl. S2), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.E.; Hanley, E.N., Jr. The musculoskeletal effects of smoking. J. Am. Acad. Orthop. Surg. 2001, 9, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Sloan, A.; Hussain, I.; Maqsood, M.; Eremin, O.; El-Sheemy, M. The effects of smoking on fracture healing. Surgeon 2010, 8, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Bydon, M.; De la Garza-Ramos, R.; Abt, N.B.; Gokaslan, Z.L.; Wolinsky, J.P.; Sciubba, D.M.; Bydon, A.; Witham, T.F. Impact of smoking on complication and pseudarthrosis rates after single- and 2-level posterolateral fusion of the lumbar spine. Spine 2014, 39, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Glassman, S.D.; Anagnost, S.C.; Parker, A.; Burke, D.; Johnson, J.R.; Dimar, J.R. The effect of cigarette smoking and smoking cessation on spinal fusion. Spine 2000, 25, 2608–2615. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Djordjevic, M.V.; Hoffmann, I. The changing cigarette. Prev. Med. 1997, 26, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Rothem, D.E.; Rothem, L.; Soudry, M.; Dahan, A.; Eliakim, R. Nicotine modulates bone metabolism-associated gene expression in osteoblast cells. J. Bone Miner. Metab. 2009, 27, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Leow, Y.H.; Maibach, H.I. Cigarette smoking, cutaneous vasculature, and tissue oxygen. Clin. Dermatol. 1998, 16, 579–584. [Google Scholar] [CrossRef]
- Lee, L.L.; Lee, J.S.; Waldman, S.D.; Casper, R.F.; Grynpas, M.D. Polycyclic aromatic hydrocarbons present in cigarette smoke cause bone loss in an ovariectomized rat model. Bone 2002, 30, 917–923. [Google Scholar] [CrossRef]
- Kung, M.H.; Yukata, K.; O’Keefe, R.J.; Zuscik, M.J. Aryl hydrocarbon receptor-mediated impairment of chondrogenesis and fracture healing by cigarette smoke and benzo(a)pyrene. J. Cell. Physiol. 2012, 227, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.; Weiner, J.A.; Chun, D.S.; Yun, J.; Cook, R.W.; Schallmo, M.S.; Kannan, A.S.; Mitchell, S.M.; Freshman, R.D.; Park, C.; et al. Mechanistic insight into the effects of Aryl Hydrocarbon Receptor activation on osteogenic differentiation. Bone Rep. 2017, 6, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Jamsa, T.; Viluksela, M.; Tuomisto, J.T.; Tuomisto, J.; Tuukkanen, J. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on bone in two rat strains with different aryl hydrocarbon receptor structures. J. Bone Miner. Res. 2001, 16, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Milbrath, M.O.; Wenger, Y.; Chang, C.W.; Emond, C.; Garabrant, D.; Gillespie, B.W.; Jolliet, O. Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environ. Health Perspect. 2009, 117, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Boutros, P.C.; Yan, R.; Moffat, I.D.; Pohjanvirta, R.; Okey, A.B. Transcriptomic responses to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in liver: Comparison of rat and mouse. BMC Genom. 2008, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Emond, C.; Birnbaum, L.S.; DeVito, M.J. Use of a physiologically based pharmacokinetic model for rats to study the influence of body fat mass and induction of CYP1A2 on the pharmacokinetics of TCDD. Environ. Health Perspect. 2006, 114, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Byard, J.L. The toxicological significance of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds in human adipose tissue. J. Toxicol. Environ. Health 1987, 22, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Hestermann, E.V.; Brown, M. Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Mol. Cell. Biol. 2003, 23, 7920–7925. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Krishnan, V.; Safe, S. Mechanism of action of alpha-naphthoflavone as an Ah receptor antagonist in MCF-7 human breast cancer cells. Toxicol. Appl. Pharmacol. 1993, 120, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.S.; Mulligan Tuttle, A.M.; Casper, R.F.; Lagunov, A.; Foster, W.G. Aryl hydrocarbon receptor antagonists attenuate the deleterious effects of benzo[a]pyrene on isolated rat follicle development. Reprod. Biomed. Online 2010, 21, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.L.; Sonn, K.; Kannan, A.; Bellary, S.; Yun, C.; Hashmi, S.; Nelson, J.; Mendoza, M.; Nickoli, M.; Ghodasra, J.; et al. Dioxin Exposure Impairs BMP-2-Mediated Spinal Fusion in a Rat Arthrodesis Model. J. Bone Jt. Surg. Am. Vol. 2015, 97, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Benayahu, D.; Shur, I.; Marom, R.; Meller, I.; Issakov, J. Cellular and molecular properties associated with osteosarcoma cells. J. Cell. Biochem. 2001, 84, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Jukkola, A.; Risteli, L.; Melkko, J.; Risteli, J. Procollagen synthesis and extracellular matrix deposition in MG-63 osteosarcoma cells. J. Bone Miner. Res. 1993, 8, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J.; Hay, E.; Saidak, Z. Integrin and cadherin signaling in bone: Role and potential therapeutic targets. Trends Endocrinol. Metab. 2014, 25, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist Annette, S.P. Chemokines and Bone. Clin. Rev. Bone Miner. Metab. 2015, 13, 61–82. [Google Scholar] [CrossRef]
- Kawakami, Y.; Ii, M.; Matsumoto, T.; Kuroda, R.; Kuroda, T.; Kwon, S.M.; Kawamoto, A.; Akimaru, H.; Mifune, Y.; Shoji, T.; et al. SDF-1/CXCR4 axis in Tie2-lineage cells including endothelial progenitor cells contributes to bone fracture healing. J. Bone Miner. Res. 2015, 30, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Yellowley, C. CXCL12/CXCR4 signaling and other recruitment and homing pathways in fracture repair. Bonekey Rep. 2013, 2, 300. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP kinases and cell migration. J. Cell Sci. 2004, 117, 4619–4628. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L. Exchange of actin subunits at the leading edge of living fibroblasts: Possible role of treadmilling. J. Cell Biol. 1985, 101, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F.; Harris, S.E.; Rosser, J.; Dallas, M.R.; Dallas, S.L.; Camacho, N.P.; Boyan, B.; Boskey, A. von Kossa staining alone is not sufficient to confirm that mineralization in vitro represents bone formation. Calcif. Tissue Int. 2003, 72, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.N.; Masica, D.L.; Gray, J.J.; McKee, M.D. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J. Bone Miner. Res. 2010, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Hadley, M.N.; Reddy, S.V. Smoking and the human vertebral column: A review of the impact of cigarette use on vertebral bone metabolism and spinal fusion. Neurosurgery 1997, 41, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Hernigou, J.; Schuind, F. Smoking as a predictor of negative outcome in diaphyseal fracture healing. Int. Orthop. 2013, 37, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Hilibrand, A.S.; Fye, M.A.; Emery, S.E.; Palumbo, M.A.; Bohlman, H.H. Impact of smoking on the outcome of anterior cervical arthrodesis with interbody or strut-grafting. J. Bone Jt. Surg. Am. Vol. 2001, 83, 668–673. [Google Scholar] [CrossRef]
- Lind, P.M.; Wejheden, C.; Lundberg, R.; Alvarez-Lloret, P.; Hermsen, S.A.; Rodriguez-Navarro, A.B.; Larsson, S.; Rannug, A. Short-term exposure to dioxin impairs bone tissue in male rats. Chemosphere 2009, 75, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Korkalainen, M.; Kallio, E.; Olkku, A.; Nelo, K.; Ilvesaro, J.; Tuukkanen, J.; Mahonen, A.; Viluksela, M. Dioxins interfere with differentiation of osteoblasts and osteoclasts. Bone 2009, 44, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Carpi, D.; Korkalainen, M.; Airoldi, L.; Fanelli, R.; Hakansson, H.; Muhonen, V.; Tuukkanen, J.; Viluksela, M.; Pastorelli, R. Dioxin-sensitive proteins in differentiating osteoblasts: Effects on bone formation in vitro. Toxicol. Sci. 2009, 108, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhang, L.; Wei, K.; Zhao, J.; Wang, Y.; Jin, F.; Xuan, K. Exposure to a continuous low dose of tetrachlorodibenzo-p-dioxin impairs the development of the tooth root in lactational rats and alters the function of apical papilla-derived stem cells. Arch. Oral Biol. 2015, 60, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.S. Regulation of bone-renal mineral and energy metabolism: The PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 61–86. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.S.; Garrett, I.R.; Schwarz, P.M.; Carnes, D.L.; Lafer, E.M.; Mundy, G.R.; Gutierrez, G.E. Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: A model for impaired mineralization in X-linked rickets (HYP). Bone 2005, 36, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M.; Kasai, A. Cigarette smoke as a trigger for the dioxin receptor-mediated signaling pathway. Cancer Lett. 2007, 252, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, U.P.; Grizzle, W.E.; Lillard, J.W., Jr. CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab. Investig. 2004, 84, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.T.; Aylward, L.L. Human response to dioxin: Aryl hydrocarbon receptor (AhR) molecular structure, function, and dose-response data for enzyme induction indicate an impaired human AhR. J. Toxicol. Environ. Health Part B Crit. Rev. 2006, 9, 147–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Liu, Y.; Maye, P.; Rowe, D.W. Examination of mineralized nodule formation in living osteoblastic cultures using fluorescent dyes. Biotechnol. Prog. 2006, 22, 1697–1701. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA | Sequences 5′-3′ | |
|---|---|---|
| CYP1A1 | Forward | AAA CCC AGC TGA CTT CAT CC |
| Reverse | TGC TCC TTG ACC ATC TTC TG | |
| RUNX2 | Forward | GGT TAA TCT CCG CAG GTC ACT |
| Reverse | CAC TGT GCT GAA GAG GCT GTT | |
| ALP | Forward | CCA TTC CCA CGT CTT CAC AT |
| Reverse | GCT TCT TGT CTG TGT CAC TCA | |
| CXCL12 | Forward | TGC CAG AGC CAA CGT CAA G |
| Reverse | CAG CCG GGC TAC AAT CTG AA | |
| CXCR4 | Forward | AGC AGG TAG CAA AGT GAC G |
| Reverse | CCT CGG TGT AGT TAT CTG AAG TG | |
| PHEX | Forward | GAG CTC AAG TTA TGC TCA TGT GAG GTG |
| Reverse | AAA TAA GAG CTC CAG AGT CGA CAG GAG | |
| OCN | Forward | TCA CAC TCC TCG CCC TAT TG |
| Reverse | TCG CTG CCC TCC TGC TTG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, C.; Katchko, K.M.; Schallmo, M.S.; Jeong, S.; Yun, J.; Chen, C.H.; Weiner, J.A.; Park, C.; George, A.; Stupp, S.I.; et al. Aryl Hydrocarbon Receptor Antagonists Mitigate the Effects of Dioxin on Critical Cellular Functions in Differentiating Human Osteoblast-Like Cells. Int. J. Mol. Sci. 2018, 19, 225. https://doi.org/10.3390/ijms19010225
Yun C, Katchko KM, Schallmo MS, Jeong S, Yun J, Chen CH, Weiner JA, Park C, George A, Stupp SI, et al. Aryl Hydrocarbon Receptor Antagonists Mitigate the Effects of Dioxin on Critical Cellular Functions in Differentiating Human Osteoblast-Like Cells. International Journal of Molecular Sciences. 2018; 19(1):225. https://doi.org/10.3390/ijms19010225
Chicago/Turabian StyleYun, Chawon, Karina M. Katchko, Michael S. Schallmo, Soyeon Jeong, Jonghwa Yun, Charlotte H. Chen, Joseph A. Weiner, Christian Park, Andrew George, Samuel I. Stupp, and et al. 2018. "Aryl Hydrocarbon Receptor Antagonists Mitigate the Effects of Dioxin on Critical Cellular Functions in Differentiating Human Osteoblast-Like Cells" International Journal of Molecular Sciences 19, no. 1: 225. https://doi.org/10.3390/ijms19010225
APA StyleYun, C., Katchko, K. M., Schallmo, M. S., Jeong, S., Yun, J., Chen, C. H., Weiner, J. A., Park, C., George, A., Stupp, S. I., Hsu, W. K., & Hsu, E. L. (2018). Aryl Hydrocarbon Receptor Antagonists Mitigate the Effects of Dioxin on Critical Cellular Functions in Differentiating Human Osteoblast-Like Cells. International Journal of Molecular Sciences, 19(1), 225. https://doi.org/10.3390/ijms19010225