Sexually Dimorphic Outcomes after Neonatal Stroke and Hypoxia-Ischemia


Abstract
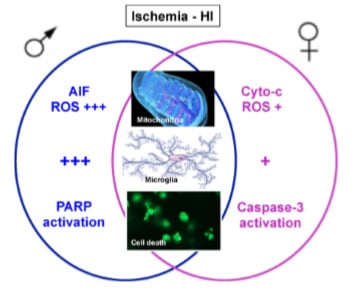
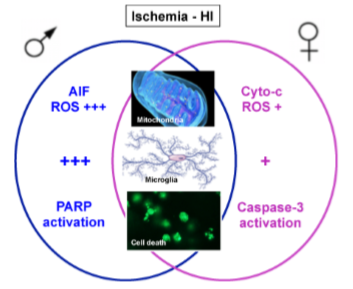
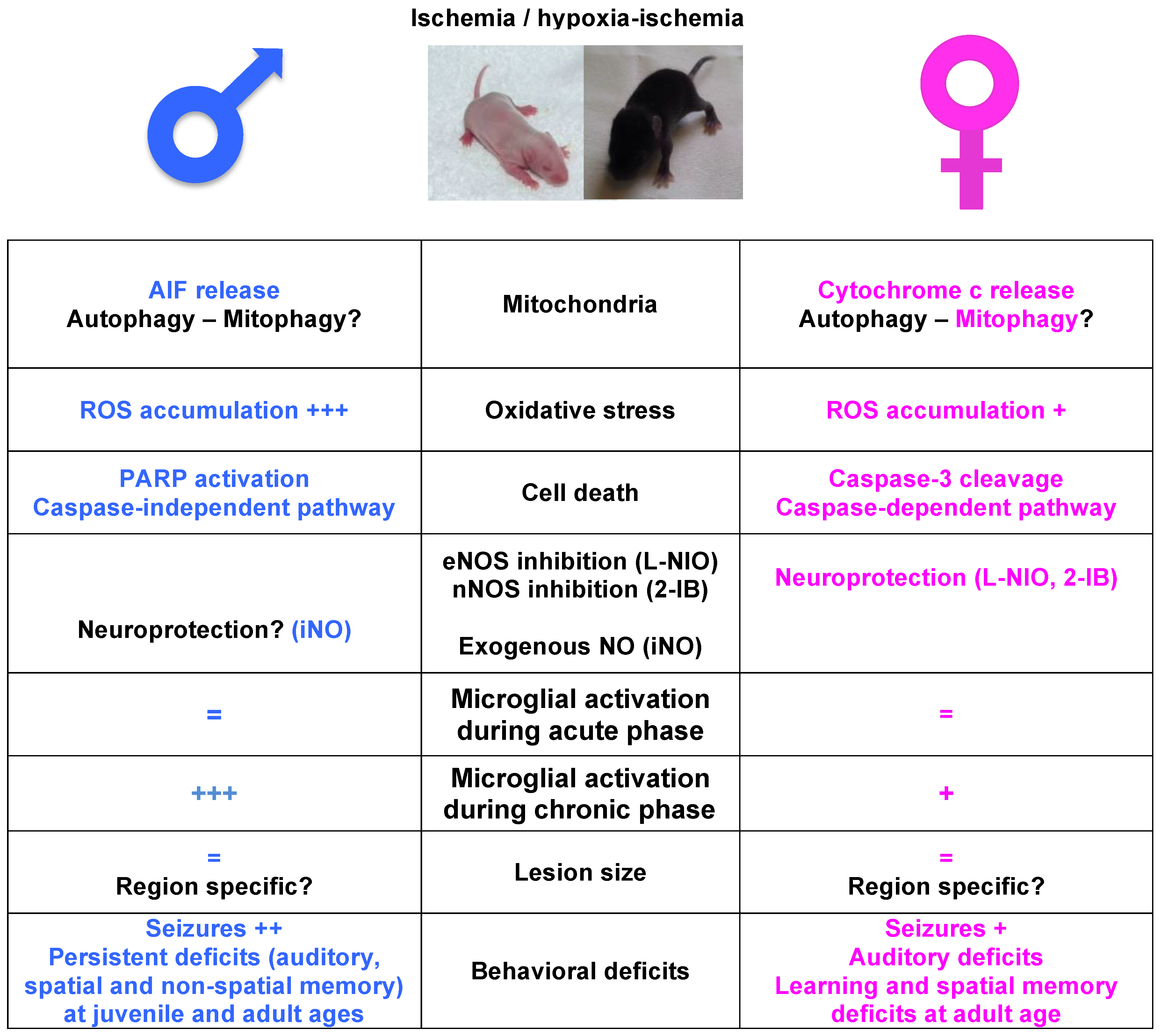
:
{kind=link}
{kind=link}
1. Introduction
2. Preclinical Models of Brain Injury and Sexual Dimorphism
3. Sexual Dimorphism in Cell Death Pathways
4. Sexual Dimorphism in Oxidative Stress
5. Sexual Dimorphism and Microglial Activation
6. Sex Dimorphism and Gonadal Hormones
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Badve, C.A.; Khanna, P.C.; Ishak, G.E. Neonatal ischemic brain injury: What every radiologist needs to know. Pediatr. Radiol. 2012, 42, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.L.; Cowan, F.M. Neonatal arterial ischaemic stroke: Obstetric issues. Semin. Fetal Neonatal Med. 2009, 14, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Tsze, D.S.; Valente, J.H. Pediatric stroke: A review. Emerg. Med. Int. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Coq, J.O.; Delcour, M.; Massicotte, V.S.; Baud, O.; Barbe, M.F. Prenatal ischemia deteriorates white matter, brain organization, and function: Implications for prematurity and cerebral palsy. Dev. Med. Child Neurol. 2016, 58 (Suppl. S4), 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kirton, A.; Armstrong-Wells, J.; Chang, T.; Deveber, G.; Rivkin, M.J.; Hernandez, M.; Carpenter, J.; Yager, J.Y.; Lynch, J.K.; Ferriero, D.M.; et al. International Pediatric Stroke Study Investigators. Symptomatic neonatal arterial ischemic stroke: The International Pediatric Stroke Study. Pediatrics 2011, 128, e1402–e1410. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Pham, T.N.; Danielson, B.; Towner, D.; Smith, L.; Johnston, S.C. Nighttime delivery and risk of neonatal encephalopathy. Am. J. Obstet. Gynecol. 2011, 204, 37.e1–37.e6. [Google Scholar] [CrossRef] [PubMed]
- Golomb, M.R. Outcomes of perinatal arterial ischemic stroke and cerebral sinovenous thrombosis. Semin. Fetal Neonatal Med. 2009, 14, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Golomb, M.R.; Dick, P.T.; MacGregor, D.L.; Curtis, R.; Sofronas, M.; deVeber, G.A. Neonatal arterial ischemic stroke and cerebral sinovenous thrombosis are more commonly diagnosed in boys. J. Child Neurol. 2004, 19, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.A.; Fitch, R.H. Sex differences in mechanisms and outcome of neonatal hypoxia-ischemia in rodent models: Implications for sex-specific neuroprotection in clinical neonatal practice. Neurol. Res. Int. 2012, 2012, 867531. [Google Scholar] [CrossRef] [PubMed]
- Gur, R.C.; Mozley, P.D.; Resnick, S. Gender differences in age effect on brain atrophy measured by magnetic resonance imaging. Proc. Natl. Acad. Sci. USA 1991, 88, 2845–2849. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; McCarthy, M.M. A starring role for microglia in brain sex differences. Neuroscientist 2015, 21, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Clancy, B.; Kersh, B.; Hyde, J.; Darlington, R.B.; Anand, K.J.; Finlay, B.L. Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics 2007, 5, 79–94. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mallard, C.; Vexler, Z.S. Modeling Ischemia in the Immature Brain: How Translational Are Animal Models? Stroke 2015, 46, 3006–3011. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.E., III; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Derugin, N.; Dingman, A.; Wendland, M.F.; Fox, C.; Bollen, A.; Vexler, Z.S. Magnetic resonance imaging as a surrogate measure for histological sub-chronic endpoint in a neonatal rat stroke model. Brain Res. 2005, 1066, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, M.; Tsuji, M.; Taguchi, A.; Kasahara, Y.; Ikeda, T. Cerebral blood flow during reperfusion predicts later brain damage in a mouse and a rat model of neonatal hypoxic-ischemic encephalopathy. Exp. Neurol. 2012, 233, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M.; Ohshima, M.; Taguchi, A.; Kasahara, Y.; Ikeda, T.; Matsuyama, T. A novel reproducible model of neonatal stroke in mice: Comparison with a hypoxia-ischemia model. Exp. Neurol. 2013, 247, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Sedik, C.; Ferriero, D.M. Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res. 1998, 810, 114–122. [Google Scholar] [CrossRef]
- Renolleau, S.; Aggoun-Zouaoui, D.; Ben-Ari, Y.; Charriaut-Marlangue, C. A model of transient unilateral focal ischemia with reperfusion in the P7 neonatal rat: Morphological changes indicative of apoptosis. Stroke 1998, 29, 1454–1460; discussion 1461. [Google Scholar] [CrossRef] [PubMed]
- Mitsufuji, N.; Yoshioka, H.; Okano, S.; Nishiki, T.; Sawada, T. A new model of transient cerebral ischemia in neonatal rats. J. Cereb. Blood Flow Metab. 1996, 16, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Leger, P.L.; Bonnin, P.; Moretti, R.; Tanaka, S.; Duranteau, J.; Renolleau, S.; Baud, O.; Charriaut-Marlangue, C. Early Recruitment of Cerebral Microcirculation by Neuronal Nitric Oxide Synthase Inhibition in a Juvenile Ischemic Rat Model. Cerebrovasc. Dis. 2016, 41, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Derugin, N.; Ferriero, D.M.; Vexler, Z.S. Neonatal reversible focal cerebral ischemia: A new model. Neurosci. Res. 1998, 32, 349–353. [Google Scholar] [CrossRef]
- Bonnin, P.; Leger, P.L.; Deroide, N.; Fau, S.; Baud, O.; Pocard, M.; Charriaut-Marlangue, C.; Renolleau, S. Impact of intracranial blood-flow redistribution on stroke size during ischemia-reperfusion in 7-day-old rats. J. Neurosci. Methods 2011, 198, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Leger, P.-L.; Besson, V.C.; Csaba, Z.; Pansiot, J.; Di Criscio, L.; Gentili, A.; Titomanlio, L.; Bonnin, P.; Baud, O.; et al. Sildenafil, a cyclic GMP phosphodiesterase inhibitor, induces microglial modulation after focal ischemia in the neonatal mouse brain. J. Neuroinflamm. 2016, 13, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelot, A.; Villapol, S.; Billette de Villemeur, T.; Renolleau, S.; Charriaut-Marlangue, C. Astrocytic demise in the developing rat and human brain after hypoxic-ischemic damage. Dev. Neurosci. 2009, 31, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Wilson, M.A.; Matsushita, H.; Zhu, C.; Lange, M.; Gustavsson, M.; Poitras, M.F.; Dawson, T.M.; Dawson, V.L.; Northington, F.; et al. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J. Neurochem. 2004, 90, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Alexander, M.; Rosenkrantz, T.S.; Sadek, M.L.; Fitch, R.H. Sex differences in behavioral outcome following neonatal hypoxia ischemia: Insights from a clinical meta-analysis and a rodent model of induced hypoxic ischemic brain injury. Exp. Neurol. 2014, 254, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Sanches, E.F.; Arteni, N.; Nicola, F.; Aristimunha, D.; Netto, C.A. Sexual dimorphism and brain lateralization impact behavioral and histological outcomes following hypoxia-ischemia in P3 and P7 rats. Neuroscience 2015, 290, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Sanches, E.F.; Arteni, N.; Nicola, F.; Boisserand, L.; Willborn, S.; Netto, C.A. Early hypoxia-ischemia causes hemisphere and sex-dependent cognitive impairment and histological damage. Neuroscience 2013, 237, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Z.; Wen, X.H.; Liu, H. Sex differences in brain MRI abnormalities and neurodevelopmental outcomes in a rat model of neonatal hypoxia-ischemia. Int. J. Neurosci. 2016, 126, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, S.R.; Omar, G.; Penn, A.A. Sex differences in a hypoxia model of preterm brain damage. Pediatr. Res. 2009, 66, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Ritzel, R.; Xu, Y.; McCullough, L.D.; Liu, F. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. J. Neuroinflamm. 2015, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.R.; Liu, C.L.; Ouyang, Y.; Blomgren, K.; Siesjo, B.K. Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J. Cereb. Blood Flow Metab. 2000, 20, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Lesuisse, C.; Martin, L.J. Immature and mature cortical neurons engage different apoptotic mechanisms involving caspase-3 and the mitogen-activated protein kinase pathway. J. Cereb. Blood Flow Metab. 2002, 22, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Renolleau, S.; Fau, S.; Charriaut-Marlangue, C. Gender-related differences in apoptotic pathways after neonatal cerebral ischemia. Neuroscientist 2008, 14, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Renolleau, S.; Fau, S.; Goyenvalle, C.; Joly, L.-M.; Chauvier, D.; Jacotot, E.; Mariani, J.; Charriaut-Marlangue, C. Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: A role for gender. J. Neurochem. 2007, 100, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Puyal, J.; Vaslin, A.; Mottier, V.; Clarke, P.G. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann. Neurol. 2009, 66, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.N.; Pettenuzzo, L.F.; Krolow, R.; Valentim, L.M.; Mota, C.S.; Dalmaz, C.; Wyse, A.T.; Netto, C.A. Autophagy in the brain of neonates following hypoxia-ischemia shows sex- and region-specific effects. Neuroscience 2014, 256, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Demarest, T.G.; Waite, E.L.; Kristian, T.; Puche, A.C.; Waddell, J.; McKenna, M.C.; Fiskum, G. Sex-dependent mitophagy and neuronal death following rat neonatal hypoxia-ischemia. Neuroscience 2016, 335, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, O.; Alvarez, A.; Revuelta, M.; Santaolalla, F.; Urtasun, A.; Hilario, E. Role of Antioxidants in Neonatal Hypoxic-Ischemic Brain Injury: New Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 265. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.N.; Pettenuzzo, L.F.; Krolow, R.; Valentim, L.M.; Mota, C.S.; Dalmaz, C.; Wyse, A.T.; Netto, C.A. Neonatal hypoxia-ischemia induces sex-related changes in rat brain mitochondria. Mitochondrion 2012, 12, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Demarest, T.G.; Schuh, R.A.; Waddell, J.; McKenna, M.C.; Fiskum, G. Sex-dependent mitochondrial respiratory impairment and oxidative stress in a rat model of neonatal hypoxic-ischemic encephalopathy. J. Neurochem. 2016, 137, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Demarest, T.G.; Schuh, R.A.; Waite, E.L.; Waddell, J.; McKenna, M.C.; Fiskum, G. Sex dependent alterations in mitochondrial electron transport chain proteins following neonatal rat cerebral hypoxic-ischemia. J. Bioenerg. Biomembr. 2016, 48, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.; Vannucci, R.C.; Towfighi, J. Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr. Res. 1990, 27, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fanjul, J.; Duran Fernandez-Feijoo, C.; Lopez-Abad, M.; Lopez Ramos, M.G.; Balada Caballe, R.; Alcantara-Horillo, S.; Camprubi Camprubi, M. Neuroprotection with hypothermia and allopurinol in an animal model of hypoxic-ischemic injury: Is it a gender question? PLoS ONE 2017, 12, e0184643. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.F.; Tymafichuk, C.N.; Bigam, D.L.; Cheung, P.Y. Effects of postresuscitation N-acetylcysteine on cerebral free radical production and perfusion during reoxygenation of hypoxic newborn piglets. Pediatr. Res. 2008, 64, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Lowe, D.W.; Rollins, L.G.; Bentzley, J.; Fraser, J.L.; Martin, R.; Singh, I.; Jenkins, D. Sex-specific effects of N-acetylcysteine in neonatal rats treated with hypothermia after severe hypoxia-ischemia. Neurosci. Res. 2016, 108, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Charriaut-Marlangue, C.; Bonnin, P.; Pham, H.; Loron, G.; Leger, P.L.; Gressens, P.; Renolleau, S.; Baud, O. Nitric oxide signaling in the brain: A new target for inhaled nitric oxide? Ann. Neurol. 2013, 73, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Bonnin, P.; Leger, P.L.; Villapol, S.; Deroide, N.; Gressens, P.; Pocard, M.; Renolleau, S.; Baud, O.; Charriaut-Marlangue, C. Dual action of NO synthases on blood flow and infarct volume consecutive to neonatal focal cerebral ischemia. Exp. Neurol. 2012, 236, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; Groenendaal, F.; Kavelaars, A.; Hagberg, H.H.; van Bel, F.; Heijnen, C.J. Gender-specific neuroprotection by 2-iminobiotin after hypoxia-ischemia in the neonatal rat via a nitric oxide independent pathway. J. Cereb. Blood Flow Metab. 2007, 27, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Charriaut-Marlangue, C.; Bonnin, P.; Gharib, A.; Leger, P.-L.; Villapol, S.; Pocard, M.; Gressens, P.; Renolleau, S.; Baud, O. Inhaled nitric oxide reduces brain damage by collateral recruitment in a neonatal stroke model. Stroke 2012, 43, 3078–3084. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Sun, Y.; Gao, J.; Wang, X.; Plesnila, N.; Blomgren, K. Inhaled nitric oxide protects males but not females from neonatal mouse hypoxia-ischemia brain injury. Transl. Stroke Res. 2013, 4, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Iosif, R.E.; Ekdahl, C.; Ahlenius, H.; Pronk, C.J.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.; Lindvall, O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef] [PubMed]
- Faustino, J.V.; Wang, X.; Jonhson, C.; Klibanov, A.; Derugin, N.; Wendland, M.; Vexler, Z.S. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Sholar, P.W.; Bilbo, S.D. Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 2012, 120, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J. Neuroinflamm. 2009, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J. Neurosci. Res. 2013, 91, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Loram, L.C.; Sholar, P.W.; Taylor, F.R.; Wiesler, J.L.; Babb, J.; Strand, K.A.; Berkelhammer, D.; Day, H.E.W.; Maier, S.F.; Watkins, L.R. Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrinology 2012, 37, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Acaz-Fonseca, E.; Duran, J.C.; Carrero, P.; Garcia-Segura, L.M.; Angeles Arevalo, M. Sex differences in glia reactivity after cortical brain injury. Glia 2015, 63, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Venna, V.R.; Weston, G.; Benashski, S.E.; Tarabishy, S.; Liu, F.; Li, J.; Conti, L.H.; McCullough, L.D. NF-kappaB contributes to the detrimental effects of social isolation after experimental stroke. Acta Neuropathol. 2012, 124, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Turtzo, L.C.; McCullough, L.D. Sex differences in stroke. Cerebrovasc. Dis. 2008, 26, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Koellhoffer, E.C.; McCullough, L.D. The effects of estrogen in ischemic stroke. Transl. Stroke Res. 2013, 4, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, S.; Moriwake, T.; Tanaka, H.; Inoue, M.; Kuo, T.; Suzuki, S.; Kanzakili, S.; Seino, Y. An ultrasensitive assay revealed age-related changes in serum oestradiol at low concentrations in both sexes from infancy to puberty. Clin. Endocrinol. 2001, 55, 789–795. [Google Scholar] [CrossRef]
- Corbier, P.; Edwards, D.A.; Roffi, J. The neonatal testosterone surge: A comparative study. Arch. Int. Physiol. Biochim. Biophys. 1992, 100, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Del Bigio, M.R.; Becker, L.E. Microglial aggregation in the dentate gyrus: A marker of mild hypoxic-ischaemic brain insult in human infants. Neuropathol. Appl. Neurobiol. 1994, 20, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Ritzel, R.; McCullough, L.D.; Liu, F. Microglia and ischemic stroke: A double-edged sword. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 73–90. [Google Scholar] [PubMed]
- Tapia-Gonzalez, S.; Carrero, P.; Pernia, O.; Garcia-Segura, L.M.; Diz-Chaves, Y. Selective oestrogen receptor (ER) modulators reduce microglia reactivity in vivo after peripheral inflammation: Potential role of microglial ERs. J. Endocrinol. 2008, 198, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Keeling, J.L.; Keller, J.N.; Huang, F.F.; Camondola, S.; Mattson, M.P. Antiinflammatory effects of estrogen on microglial activation. Endocrinology 2000, 141, 3646–3656. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; McArthur, S. Oestrogen and immunomodulation: New mechanisms that impact on peripheral and central immunity. Curr. Opin. Pharmacol. 2013, 13, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Pansiot, J.; Pham, H.; Dalous, J.; Chevenne, D.; Colella, M.; Schwendimann, L.; Fafouri, A.; Mairesse, J.; Moretti, R.; Schang, A.-L.; et al. Glial response to 17beta-estradiol in neonatal rats with excitotoxic brain injury. Exp. Neurol. 2016, 282, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, P.; Slowik, A.; Zendedel, A.; Johann, S.; Dang, J.; Beyer, C. Regulation of hypoxia-induced inflammatory responses and M1-M2 phenotype switch of primary rat microglia by sex steroids. J. Mol. Neurosci. 2014, 52, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Favrais, G.; van de Looij, Y.; Fleiss, B.; Ramanantsoa, N.; Bonnin, P.; Stoltenburg-Didinger, G.; Lacaud, A.; Saliba, E.; Dammann, O.; Gallego, J.; et al. Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 2011, 70, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, M.D.; Xia, Y.P.; Gao, Y.; Zhu, Y.Y.; Chen, S.C.; Mao, L.; He, Q.W.; Yue, Z.Y.; Hu, B. Microrna-130a regulates cerebral ischemia-induced blood-brain barrier permeability by targeting homeobox a5. FASEB J. 2017. [Google Scholar] [CrossRef]
- Xu, X.; Wen, Z.; Zhao, N.; Xu, X.; Wang, F.; Gao, J.; Jiang, Y.; Liu, X. MicroRNA-1906, a novel regulator of toll-like receptor 4, ameliorates ischemic injury after experimental stroke in mice. J. Neurosci. 2017, 37, 10498–10515. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stary, C.M. Targeting glial mitochondrial function for protection from cerebral ischemia: Relevance, mechanisms, and the role of micrornas. Oxid. Med. Cell. Longev. 2016, 2016, 6032306. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ma, Q.; Li, Y.; Li, B.; Zhang, L. Inhibition of microRNA-210 suppresses pro-inflammatory response and reduces acute brain injury of ischemic stroke in mice. Exp. Neurol. 2017, 300, 41–50. [Google Scholar]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charriaut-Marlangue, C.; Besson, V.C.; Baud, O. Sexually Dimorphic Outcomes after Neonatal Stroke and Hypoxia-Ischemia. Int. J. Mol. Sci. 2018, 19, 61. https://doi.org/10.3390/ijms19010061
Charriaut-Marlangue C, Besson VC, Baud O. Sexually Dimorphic Outcomes after Neonatal Stroke and Hypoxia-Ischemia. International Journal of Molecular Sciences. 2018; 19(1):61. https://doi.org/10.3390/ijms19010061
Chicago/Turabian StyleCharriaut-Marlangue, Christiane, Valérie C. Besson, and Olivier Baud. 2018. "Sexually Dimorphic Outcomes after Neonatal Stroke and Hypoxia-Ischemia" International Journal of Molecular Sciences 19, no. 1: 61. https://doi.org/10.3390/ijms19010061
APA StyleCharriaut-Marlangue, C., Besson, V. C., & Baud, O. (2018). Sexually Dimorphic Outcomes after Neonatal Stroke and Hypoxia-Ischemia. International Journal of Molecular Sciences, 19(1), 61. https://doi.org/10.3390/ijms19010061