Thiopurine Drugs Repositioned as Tyrosinase Inhibitors

Abstract
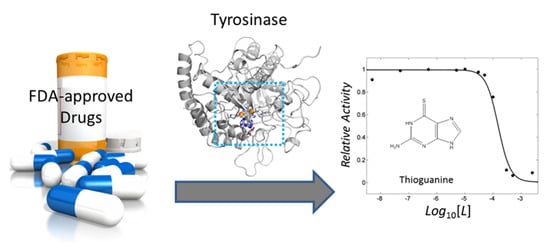
:
1. Introduction
2. Results
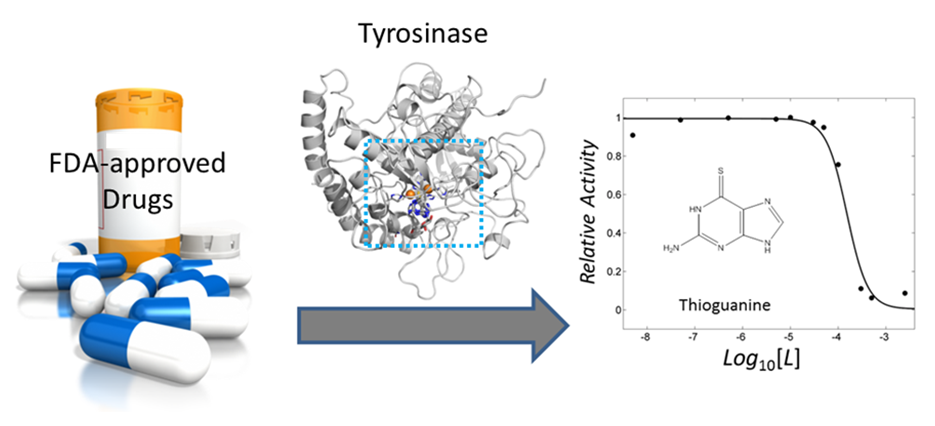
2.1. Structure-Based Docking Simulation Revealed Thioguanine as a Tyrosinase Inhibitor
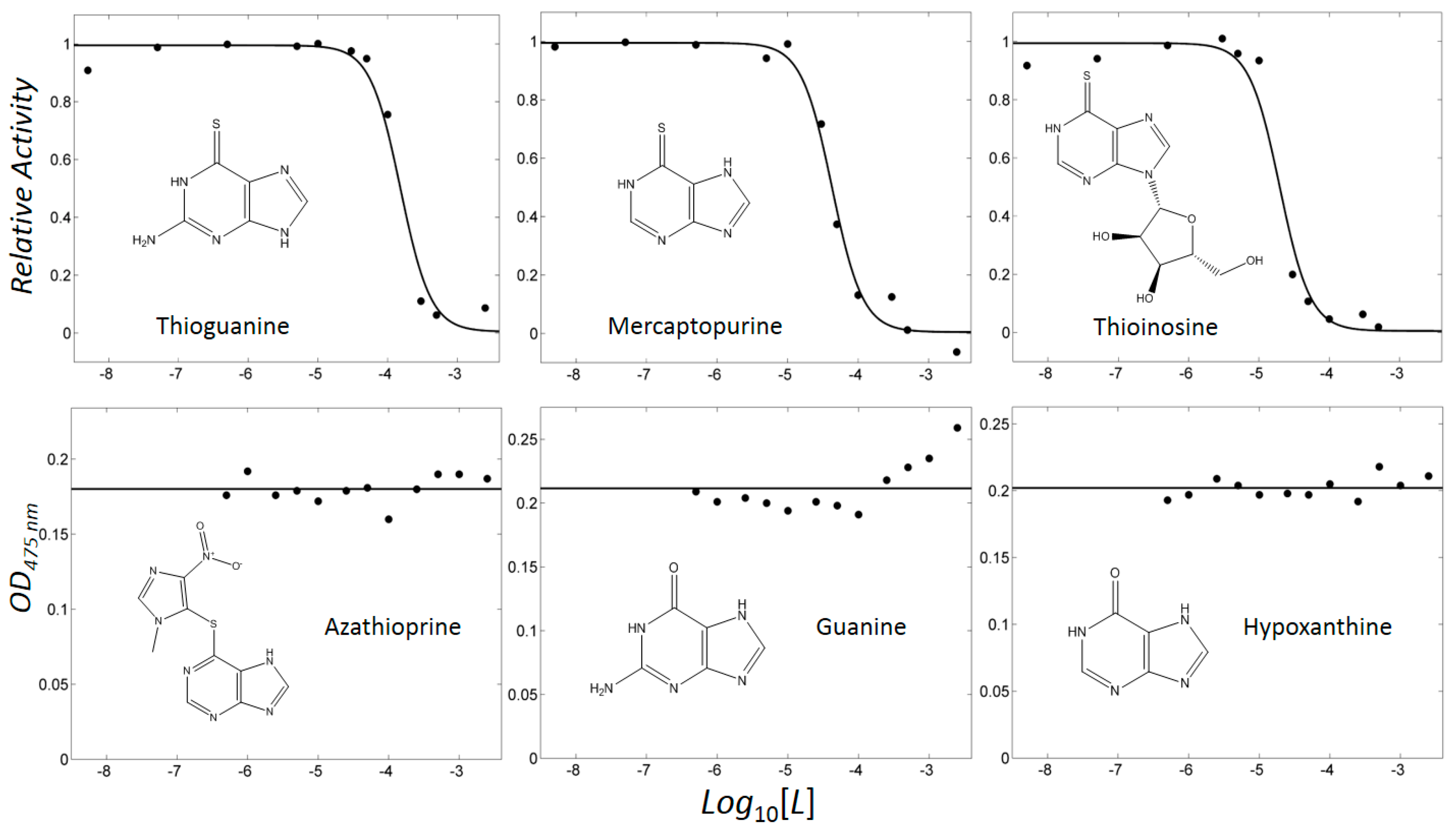
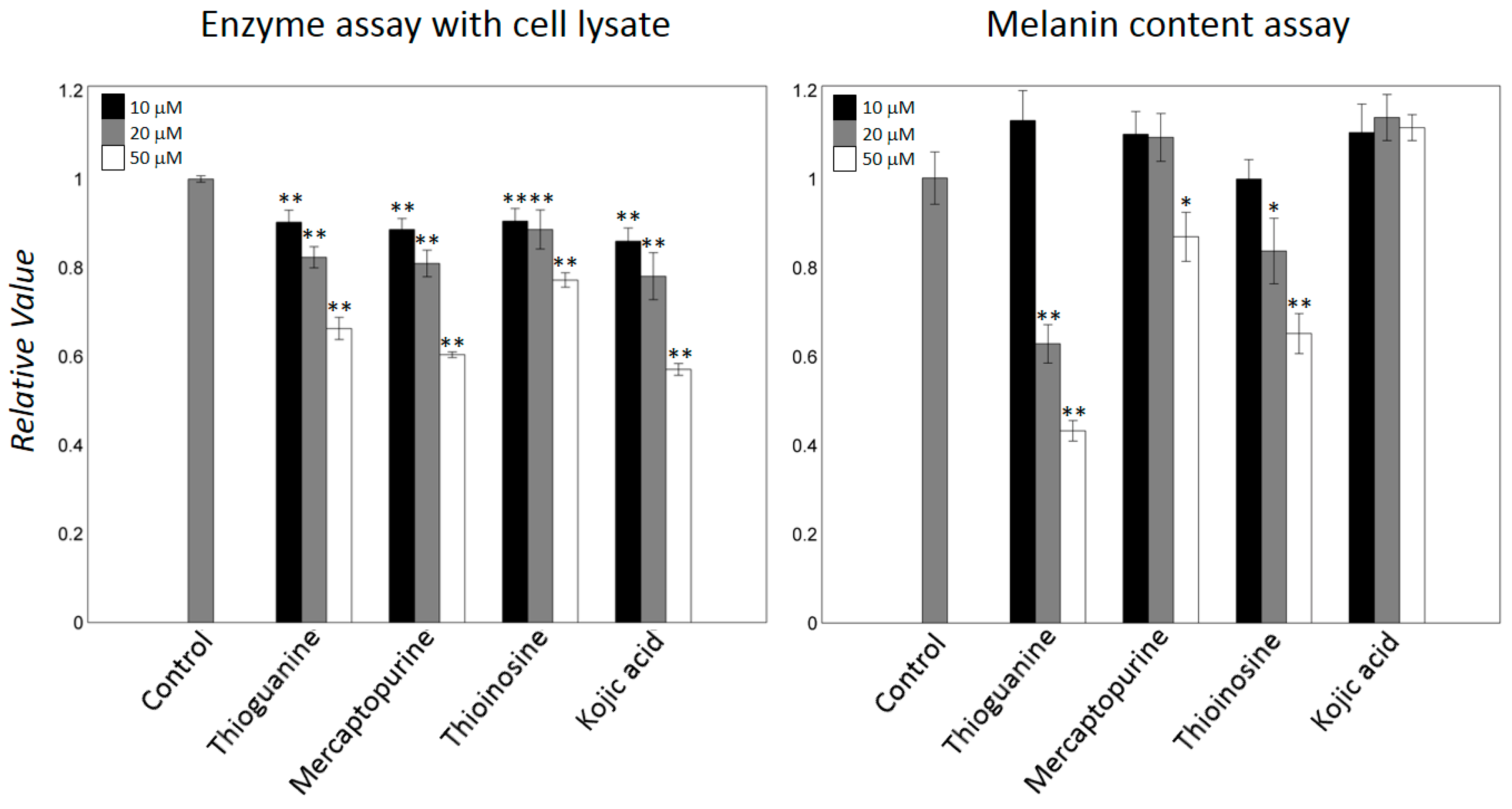
2.2. Mercaptopurine and Thioinosine Inhibited Tyrosine Activity
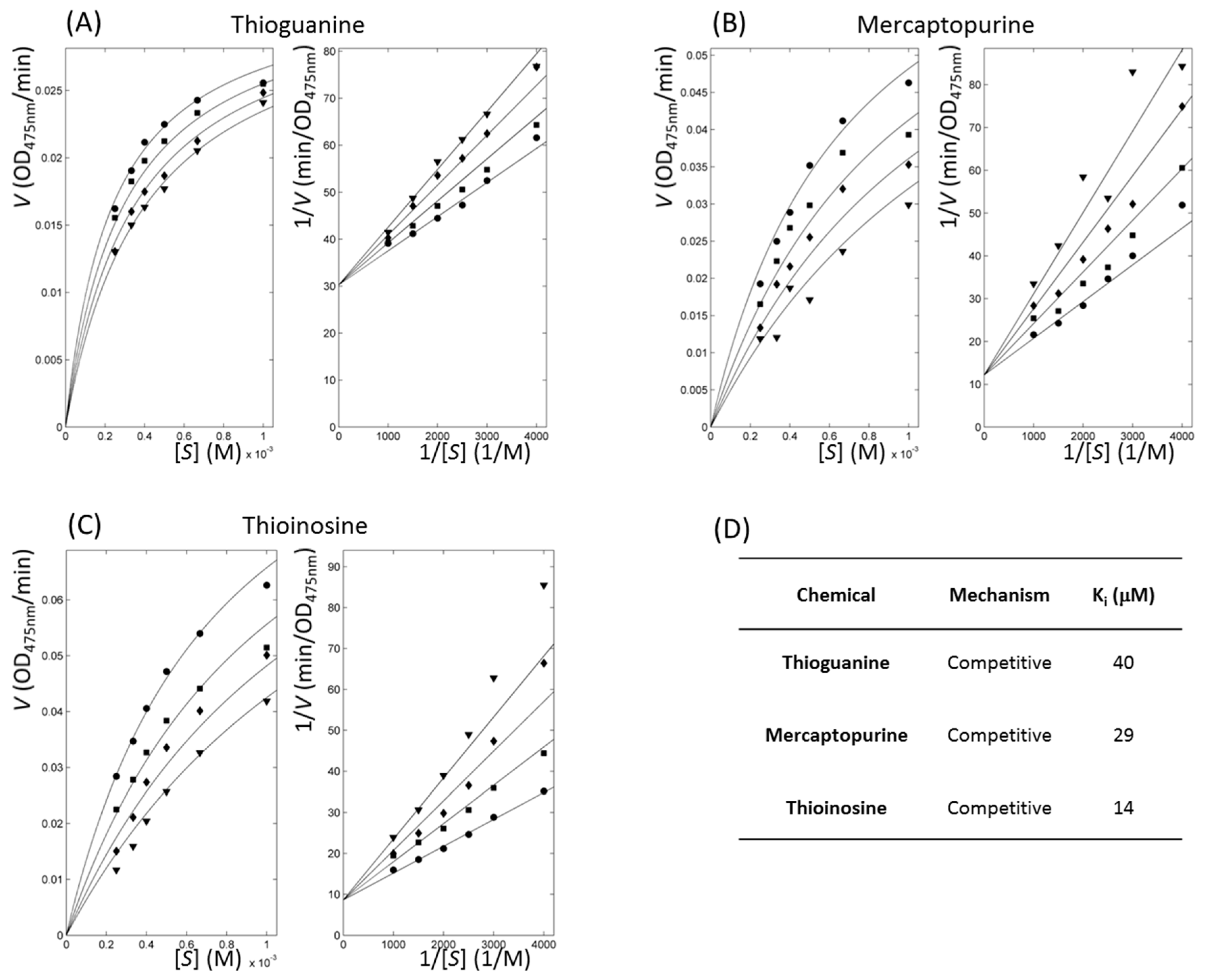
2.3. Enzyme Inhibitory Kinetics Classified Thiopurine Drugs as Competitive Inhibitors
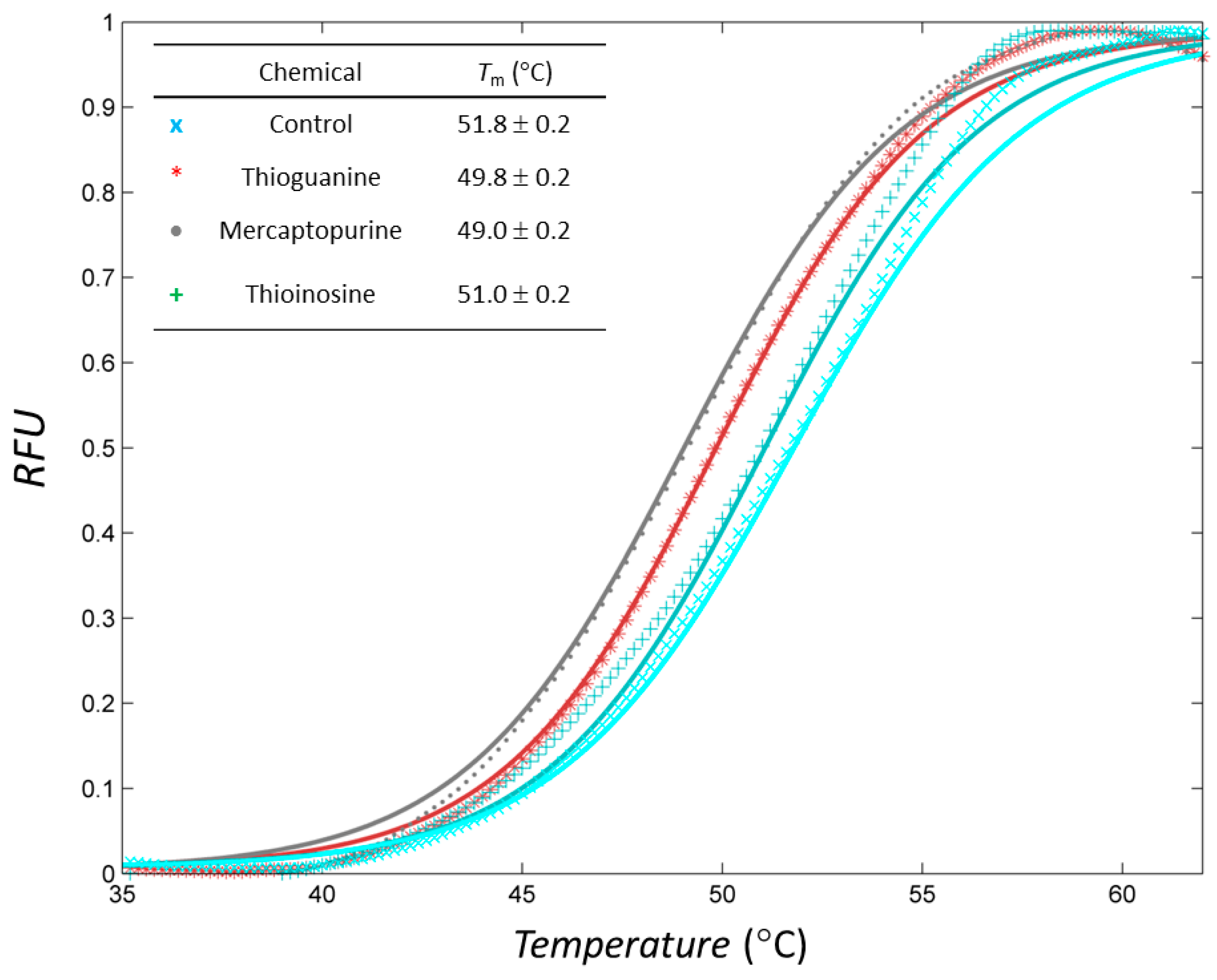
2.4. Thiopurine Drugs also Inhibited Mammalian Tyrosinase
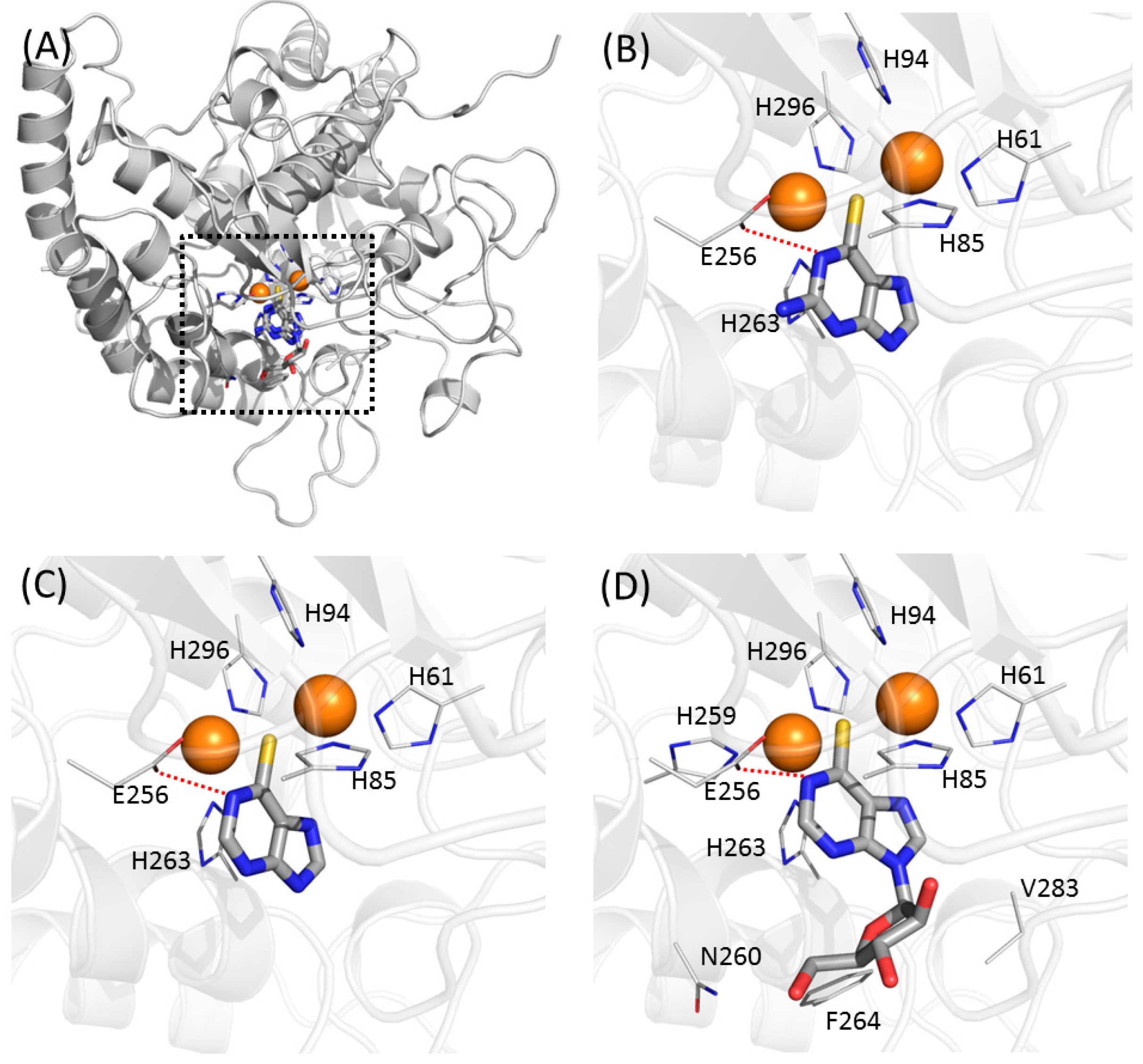
2.5. Docking Simulation Suggested Intermolecular Interactions Involved Atomic Contributions
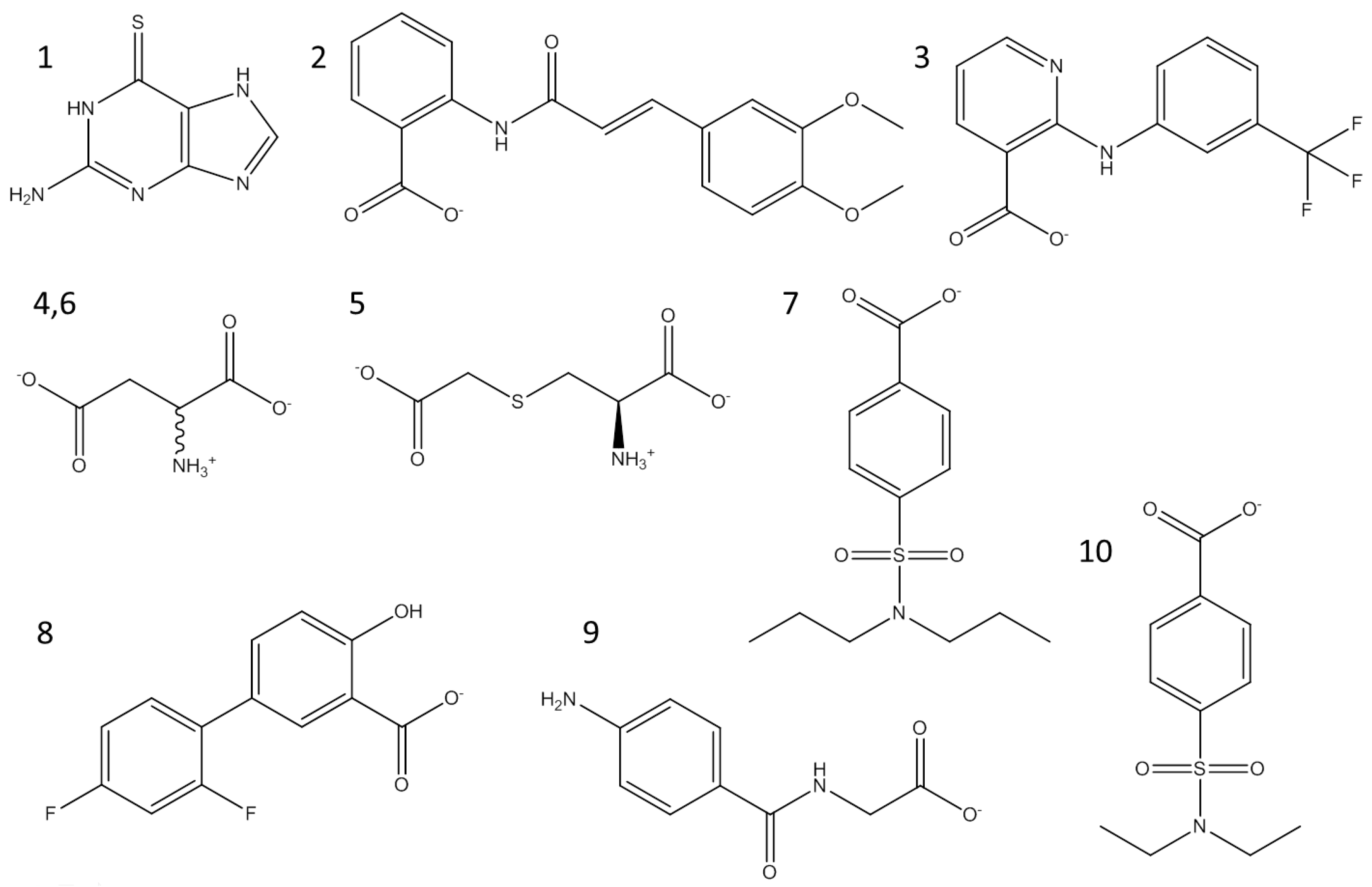
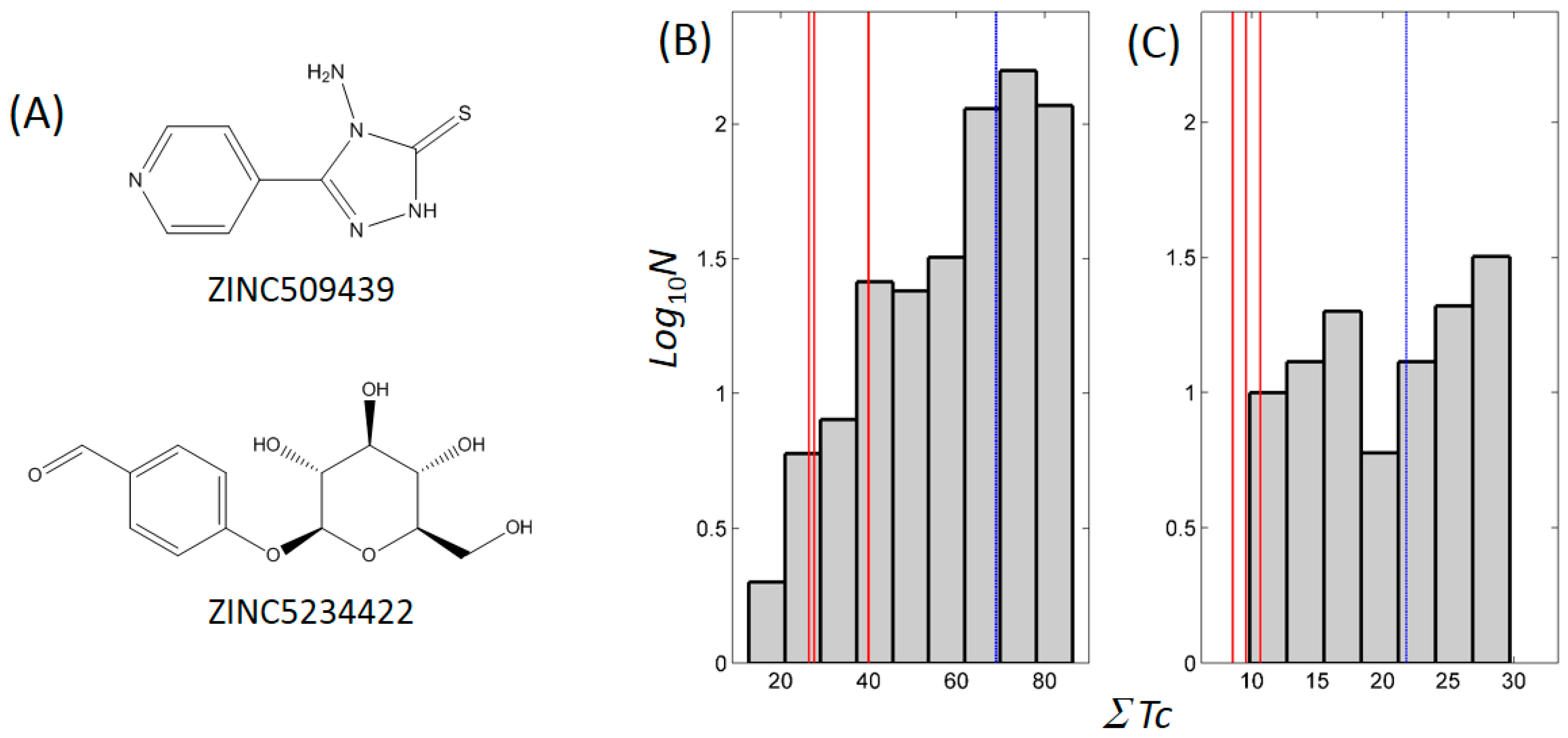
2.6. Cheminformatics Supported Novel Chemical Structures in Thiopurine Tyrosinase Inhibitors
3. Discussion
4. Materials and Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.L.; Li, K.M.; Balu, N.; Saeed, M.; Devanesan, P.; Higginbotham, S.; Zhao, J.; Gross, M.L.; Rogan, E.G. Catechol ortho-quinones: The electrophilic compounds that form depurinating DNA adducts and could initiate cancer and other diseases. Carcinogenesis 2002, 23, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Li, X.; Jankovic, J. The association between Parkinson’s disease and melanoma. Int. J. Cancer 2011, 128, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Sendoel, A.; Kohler, I.; Fellmann, C.; Lowe, S.W.; Hengartner, M.O. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 2010, 465, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, M.R.; Tundis, R.; Menichini, F. Natural and Synthetic Tyrosinase Inhibitors as Antibrowning Agents: An Update. Compr. Rev. Food Sci. Food Saf. 2012, 11, 378–398. [Google Scholar] [CrossRef]
- Khan, M.T. Novel tyrosinase inhibitors from natural resources—Their computational studies. Curr. Med. Chem. 2012, 19, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: Structure, inhibition mechanism and perspective for the future. Cell. Mol. Life Sci. 2005, 62, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.Y.; Sharma, V.K.; Sharma, N. Mushroom tyrosinase: Recent prospects. J. Agric. Food Chem. 2003, 51, 2837–2853. [Google Scholar] [CrossRef] [PubMed]
- Ubeid, A.A.; Do, S.; Nye, C.; Hantash, B.M. Potent low toxicity inhibition of human melanogenesis by novel indole-containing octapeptides. Biochim. Biophys. Acta 2012, 1820, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Solano, F.; Briganti, S.; Picardo, M.; Ghanem, G. Hypopigmenting agents: An updated review on biological, chemical and clinical aspects. Pigment Cell Melanoma Res. 2006, 19, 550–571. [Google Scholar] [CrossRef] [PubMed]
- Halaouli, S.; Asther, M.; Sigoillot, J.C.; Hamdi, M.; Lomascolo, A. Fungal tyrosinases: New prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 2006, 100, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef] [PubMed]
- Goldfeder, M.; Kanteev, M.; Isaschar-Ovdat, S.; Adir, N.; Fishman, A. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 2014, 5, 4505. [Google Scholar] [CrossRef] [PubMed]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deri, B.; Kanteev, M.; Goldfeder, M.; Lecina, D.; Guallar, V.; Adir, N.; Fishman, A. The unravelling of the complex pattern of tyrosinase inhibition. Sci. Rep. 2016, 6, 34993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klabunde, T.; Eicken, C.; Sacchettini, J.C.; Krebs, B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Mol. Biol. 1998, 5, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, A.; Oyama, T.; Takahashi, S.; Abe, H.; Kamiya, T.; Abe, T.; Tanuma, S.I. Structure-activity relationships of the thujaplicins for inhibition of human tyrosinase. Bioorg. Med. Chem. 2014, 22, 6193–6200. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Sun, W.; Gao, S.; Luan, S.; Li, D.; Chen, R.; Zhang, Q.; Chen, L.; Huang, J.; Li, H. Structure based discovery of clomifene as a potent inhibitor of cancer-associated mutant IDH1. Oncotarget 2017, 8, 44255–44265. [Google Scholar] [CrossRef] [PubMed]
- Segura-Cabrera, A.; Tripathi, R.; Zhang, X.; Gui, L.; Chou, T.F.; Komurov, K. A structure- and chemical genomics-based approach for repositioning of drugs against VCP/p97 ATPase. Sci. Rep. 2017, 7, 44912. [Google Scholar] [CrossRef] [PubMed]
- Sam, E.; Athri, P. Web-based drug repurposing tools: A survey. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- March-Vila, E.; Pinzi, L.; Sturm, N.; Tinivella, A.; Engkvist, O.; Chen, H.; Rastelli, G. On the Integration of In Silico Drug Design Methods for Drug Repurposing. Front. Pharmacol. 2017, 8, 298. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liu, L.J.; Dong, Z.Q.; Lu, L.; Wang, M.; Leung, C.H.; Ma, D.L.; Wang, Y. Structure-based discovery of an immunomodulatory inhibitor of TLR1-TLR2 heterodimerization from a natural product-like database. Chem. Commun. (Camb.) 2015, 51, 11178–11181. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.H.; Chan, D.S.; Kwan, M.H.; Cheng, Z.; Wong, C.Y.; Zhu, G.Y.; Fong, W.F.; Ma, D.L. Structure-based repurposing of FDA-approved drugs as TNF-α inhibitors. ChemMedChem 2011, 6, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Chan, D.S.; Leung, C.H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Zhang, W.; Li, L.; Xie, Y.; Li, W.; Huang, N. A new target for an old drug: Identifying mitoxantrone as a nanomolar inhibitor of PIM1 kinase via kinome-wide selectivity modeling. J. Med. Chem. 2013, 56, 2619–2629. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, S.J.; Jee, J.G. Analogues of ethionamide, a drug used for multidrug-resistant tuberculosis, exhibit potent inhibition of tyrosinase. Eur. J. Med. Chem. 2015, 106, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jee, J.G. Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors. Int. J. Mol. Sci. 2015, 16, 28534–28548. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Choi, K.E.; Park, S.J.; Kim, S.Y.; Jee, J.G. Ensemble-Based Virtual Screening Led to the Discovery of New Classes of Potent Tyrosinase Inhibitors. J. Chem. Inf. Model. 2016, 56, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Shoichet, B.K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 2010, 50, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J. Using ZINC to acquire a virtual screening library. Curr. Protoc. Bioinform. 2008. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Toriyama, M. The inhibitory effect of non-steroidal anti-inflammatory drugs (NSAIDs) on the monophenolase and diphenolase activities of mushroom tyrosinase. Int. J. Mol. Sci. 2011, 12, 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Wisansky, W.A.; Martin, G.J.; Ansbacher, S. p-aminobenzoic acid and tyrosinase activity. J. Am. Chem. Soc. 1941, 63, 1771–1772. [Google Scholar] [CrossRef]
- McGovern, S.L.; Helfand, B.T.; Feng, B.; Shoichet, B.K. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003, 46, 4265–4272. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keseru, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, A.A.; Pinto, P.A.; Fraga, I.; Bezerra, R.M.F. Diagnosis of Enzyme Inhibition Using Excel Solver: A Combined Dry and Wet Laboratory Exercise. J. Chem. Educ. 2014, 91, 1017–1021. [Google Scholar] [CrossRef]
- Martin, R.B. Disadvantages of Double Reciprocal Plots. J. Chem. Educ. 1997, 74, 1238. [Google Scholar] [CrossRef]
- Sun, W.; Wendt, M.; Klebe, G.; Rohm, K.H. On the interpretation of tyrosinase inhibition kinetics. J. Enzym. Inhib. Med. Chem. 2014, 29, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Petit, E.; Langouet, S.; Akhdar, H.; Nicolas-Nicolaz, C.; Guillouzo, A.; Morel, F. Differential toxic effects of azathioprine, 6-mercaptopurine and 6-thioguanine on human hepatocytes. Toxicol. In Vitro 2008, 22, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Oh, J.H.; Oh, I.G.; Park, C.H.; Chung, J.H. [6]-Shogaol inhibits melanogenesis in B16 mouse melanoma cells through activation of the ERK pathway. Acta Pharmacol. Sin. 2013, 34, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Hamidian, H.; Tagizadeh, R.; Fozooni, S.; Abbasalipour, V.; Taheri, A.; Namjou, M. Synthesis of novel azo compounds containing 5(4H)-oxazolone ring as potent tyrosinase inhibitors. Bioorg. Med. Chem. 2013, 21, 2088–2092. [Google Scholar] [CrossRef] [PubMed]
- Bandgar, B.P.; Adsul, L.K.; Chavan, H.V.; Shringare, S.N.; Korbad, B.L.; Jalde, S.S.; Lonikar, S.V.; Nile, S.H.; Shirfule, A.L. Synthesis, biological evaluation, and molecular docking of N-{3-[3-(9-methyl-9H-carbazol-3-yl)-acryloyl]-phenyl}-benzamide/amide derivatives as xanthine oxidase and tyrosinase inhibitors. Bioorg. Med. Chem. 2012, 20, 5649–5657. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, version 1.3r1; Schrodinger, LLC: New York, NY, USA, 2010. [Google Scholar]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Khatib, S.; Nerya, O.; Musa, R.; Tamir, S.; Peter, T.; Vaya, J. Enhanced substituted resorcinol hydrophobicity augments tyrosinase inhibition potency. J. Med. Chem. 2007, 50, 2676–2681. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Aronov, A.M.; McClain, B.; Moody, C.S.; Murcko, M.A. Kinase-likeness and kinase-privileged fragments: Toward virtual polypharmacology. J. Med. Chem. 2008, 51, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Keough, D.T.; Skinner-Adams, T.; Jones, M.K.; Ng, A.L.; Brereton, I.M.; Guddat, L.W.; de Jersey, J. Lead compounds for antimalarial chemotherapy: Purine base analogs discriminate between human and P. falciparum 6-oxopurine phosphoribosyltransferases. J. Med. Chem. 2006, 49, 7479–7486. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.F.; Wu, S.H.; Yang, Y.L.; Choong, K.F.; Chen, S.T. The screening and characterization of 6-aminopurine-based xanthine oxidase inhibitors. Bioorg. Med. Chem. 2007, 15, 3450–3456. [Google Scholar] [CrossRef] [PubMed]
- Shea, T.A.; Burburan, P.J.; Matubia, V.N.; Ramcharan, S.S.; Rosario, I., Jr.; Parkin, D.W.; Stockman, B.J. Identification of proton-pump inhibitor drugs that inhibit Trichomonas vaginalis uridine nucleoside ribohydrolase. Bioorg. Med. Chem. Lett. 2014, 24, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Santana, L.; Gonzalez-Diaz, H.; Quezada, E.; Uriarte, E.; Yanez, M.; Vina, D.; Orallo, F. Quantitative structure-activity relationship and complex network approach to monoamine oxidase A and B inhibitors. J. Med. Chem. 2008, 51, 6740–6751. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, U.F.; Majjigapu, S.R.; Chambon, M.; Bron, S.; Pilotte, L.; Colau, D.; Van den Eynde, B.J.; Turcatti, G.; Vogel, P.; Zoete, V.; et al. Detailed analysis and follow-up studies of a high-throughput screening for indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. Eur. J. Med. Chem. 2014, 84, 284–301. [Google Scholar] [CrossRef] [PubMed]
- Alijanianzadeh, M.; Saboury, A.A.; Mansuri-Torshizi, H.; Haghbeen, K.; Moosavi-Movahedi, A.A. The inhibitory effect of some new synthesized xanthates on mushroom tyrosinase activities. J. Enzym. Inhib. Med. Chem. 2007, 22, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated docking screens: A feasibility study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Motulsky, H.J.; Ransnas, L.A. Fitting curves to data using nonlinear regression: A practical and nonmathematical review. FASEB J. 1987, 1, 365–374. [Google Scholar] [PubMed]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | 2D Structure | MW | LogP & | Ki (μM) | LE # |
|---|---|---|---|---|---|
| Thioguanine |  | 167 | 0.60 | 52 | 0.49 |
| Mercaptopurine |  | 152 | 1.00 | 16 | 0.61 |
| Thioinosine |  | 284 | −0.90 | 8 | 0.34 |
| Azathioprine |  | 277 | 1.15 | >1000 | NA |
| Guanine |  | 151 | −0.77 | >1000 | NA |
| Hypoxanthine |  | 136 | −0.35 | >1000 | NA |
| Kojic acid |  | 142 | −0.16 | 13 | Control |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Lee, Y.-M.; Jee, J.-G. Thiopurine Drugs Repositioned as Tyrosinase Inhibitors. Int. J. Mol. Sci. 2018, 19, 77. https://doi.org/10.3390/ijms19010077
Choi J, Lee Y-M, Jee J-G. Thiopurine Drugs Repositioned as Tyrosinase Inhibitors. International Journal of Molecular Sciences. 2018; 19(1):77. https://doi.org/10.3390/ijms19010077
Chicago/Turabian StyleChoi, Joonhyeok, You-Mie Lee, and Jun-Goo Jee. 2018. "Thiopurine Drugs Repositioned as Tyrosinase Inhibitors" International Journal of Molecular Sciences 19, no. 1: 77. https://doi.org/10.3390/ijms19010077
APA StyleChoi, J., Lee, Y. -M., & Jee, J. -G. (2018). Thiopurine Drugs Repositioned as Tyrosinase Inhibitors. International Journal of Molecular Sciences, 19(1), 77. https://doi.org/10.3390/ijms19010077