Molecular Mechanisms of H. pylori-Induced DNA Double-Strand Breaks

Abstract
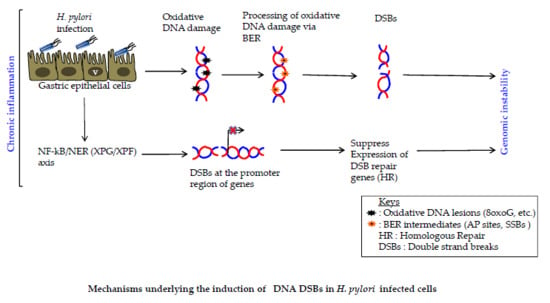
:
1. Introduction
2. H. pylori Induces Inflammation-Dependent DNA Damage
3. H. pylori Induces Base Excision Repair (BER) Intermediate-Dependent Double Strand Breaks (DSBs)
4. NF-κB-iNOS Axis-Dependent DSB Formation
5. NF-κB-Nucleotide Excision Repair (NER) Axis-Dependent DSB Formation
6. H. pylori Impairs DSBs Repair
7. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Kuper, H.; Adami, H.O.; Trichopoulos, D. Infections as a major preventable cause of human cancer. J. Int. Med. 2000, 248, 171–183. [Google Scholar] [CrossRef] [Green Version]
- De Martel, C.; Forman, D.; Plummer, M. Gastric cancer: Epidemiology and risk factors. Gastroenterol. Clin. 2013, 42, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M., Jr.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Covacci, A.; Telford, J.L.; Del Giudice, G.; Parsonnet, J.; Rappuoli, R. Helicobacter pylori virulence and genetic geography. Science 1999, 284, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Rappuoli, R. Living dangerously: How Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2001, 2, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Monack, D.M.; Mueller, A.; Falkow, S. Persistent bacterial infections: The interface of the pathogen and the host immune system. Nat. Rev. Microbiol. 2004, 2, 747–765. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First american cancer society award lecture on cancer epidemiology and prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Smoot, D.T.; Wynn, Z.; Elliott, T.B.; Allen, C.R.; Mekasha, G.; Naab, T.; Ashktorab, H. Effects of Helicobacter pylori on proliferation of gastric epithelial cells in vitro. Am. J. Gastroenterol. 1999, 94, 1508–1511. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, J.D.; Giudice, G.D.; Rappuoli, R. Helicobacter pylori interactions with host serum and extracellular matrix proteins: Potential role in the infectious process. Microbiol. Mol. Biol. Rev. 2002, 66, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Hartung, M.L.; Gruber, D.C.; Koch, K.N.; Gruter, L.; Rehrauer, H.; Tegtmeyer, N.; Backert, S.; Muller, A. H. Pylori-induced DNA strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-κB target gene expression. Cell Rep. 2015, 13, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toller, I.M.; Neelsen, K.J.; Steger, M.; Hartung, M.L.; Hottiger, M.O.; Stucki, M.; Kalali, B.; Gerhard, M.; Sartori, A.A.; Lopes, M.; et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14944–14949. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Miura, S.; Mori, M.; Kai, A.; Suzuki, H.; Fukumura, D.; Suematsu, M.; Tsuchiya, M. Rebamipide, a novel antiulcer agent, attenuates Helicobacter pylori induced gastric mucosal cell injury associated with neutrophil derived oxidants. Gut 1994, 35, 1375–1378. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, P.A. Oxy-radicals and cancer. Lancet 1994, 344, 862–863. [Google Scholar] [CrossRef]
- Feig, D.I.; Reid, T.M.; Loeb, L.A. Reactive oxygen species in tumorigenesis. Cancer Res. 1994, 54, 1890s–1894s. [Google Scholar] [PubMed]
- Schreck, R.R. Tumor suppressor gene (Rb and p53) mutations in osteosarcoma. Ped. Hematol. Oncol. 1992, 9, ix–x. [Google Scholar] [CrossRef]
- D’Angio, C.T.; Finkelstein, J.N. Oxygen regulation of gene expression: A study in opposites. Mol. Genet. MeTable 2000, 71, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Adler, V.; Yin, Z.; Tew, K.D.; Ronai, Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene 1999, 18, 6104–6111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef] [PubMed]
- Eck, M.; Schmausser, B.; Scheller, K.; Toksoy, A.; Kraus, M.; Menzel, T.; Muller-Hermelink, H.K.; Gillitzer, R. CXC chemokines Groα/IL-8 and IP-10/MIG in Helicobacter pylori gastritis. Clin. Exp. Immunol. 2000, 122, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Shimada, T.; Ohtsuka, Y.; Hiraishi, H.; Terano, A. Proinflammatory cytokines and Helicobacter pylori stimulate CC-chemokine expression in gastric epithelial cells. J. Physiol. Pharmacol. 1997, 48, 405–413. [Google Scholar] [PubMed]
- Nozawa, Y.; Nishihara, K.; Peek, R.M.; Nakano, M.; Uji, T.; Ajioka, H.; Matsuura, N.; Miyake, H. Identification of a signaling cascade for interleukin-8 production by Helicobacter pylori in human gastric epithelial cells. Biochem. Pharmacol. 2002, 64, 21–30. [Google Scholar] [CrossRef]
- Naito, Y.; Yoshikawa, T. Molecular and cellular mechanisms involved in Helicobacter pylori-induced inflammation and oxidative stress. Free Radic. Biol. Med. 2002, 33, 323–336. [Google Scholar] [CrossRef]
- Obst, B.; Wagner, S.; Sewing, K.F.; Beil, W. Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis 2000, 21, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Uchida, T.; Tsukamoto, Y.; Watada, M.; Yamaguchi, N.; Yamamoto, K.; Shiota, S.; Moriyama, M.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infect. Immun. 2014, 82, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Murphy, D.L.; Sweasy, J.B. Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during Helicobacter pylori infection. Oncogenesis 2014, 3, e128. [Google Scholar] [CrossRef] [PubMed]
- Perryman, S.V.; Sylvester, K.G. Repair and regeneration: Opportunities for carcinogenesis from tissue stem cells. J. Cell Mol. Med. 2006, 10, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A. Role of oxygen free radicals in carcinogenesis and brain ischemia. FASEB J. 1990, 4, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Du, M.Q.; Carmichael, P.L.; Phillips, D.H. Induction of activating mutations in the human c-Ha-ras-1 proto-oncogene by oxygen free radicals. Mol Carcinog 1994, 11, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Hubscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic g:C → t:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Farrington, S.M.; Tenesa, A.; Barnetson, R.; Wiltshire, A.; Prendergast, J.; Porteous, M.; Campbell, H.; Dunlop, M.G. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am. J. Hum. Genet. 2005, 77, 112–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahjabeen, I.; Masood, N.; Baig, R.M.; Sabir, M.; Inayat, U.; Malik, F.A.; Kayani, M.A. Novel mutations of OGG1 base excision repair pathway gene in laryngeal cancer patients. Fam. Cancer 2012, 11, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Tao, H.; Goto, M.; Igarashi, H.; Taniguchi, T.; Maekawa, M.; Takezaki, T.; Sugimura, H. Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer. Carcinogenesis 2004, 25, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L.; et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allinson, S.L.; Sleeth, K.M.; Matthewman, G.E.; Dianov, G.L. Orchestration of base excision repair by controlling the rates of enzymatic activities. DNA Repair 2004, 3, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef] [PubMed]
- Aburatani, H.; Hippo, Y.; Ishida, T.; Takashima, R.; Matsuba, C.; Kodama, T.; Takao, M.; Yasui, A.; Yamamoto, K.; Asano, M. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutm homologue. Cancer Res. 1997, 57, 2151–2156. [Google Scholar] [PubMed]
- Fortini, P.; Parlanti, E.; Sidorkina, O.M.; Laval, J.; Dogliotti, E. The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J. Biol. Chem. 1999, 274, 15230–15236. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S. Involvement of mammalian OGG1 (MMH) in excision of the 8-hydroxyguanine residue in DNA. Free Radic. Biol. Med. 2002, 32, 813–821. [Google Scholar] [CrossRef]
- Mokkapati, S.K.; Wiederhold, L.; Hazra, T.K.; Mitra, S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: Functional collaboration between two human DNA glycosylases. Biochemistry 2004, 43, 11596–11604. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and ap endonuclease. DNA Repair 2007, 6, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Pascucci, B.; Parlanti, E.; D’Errico, M.; Simonelli, V.; Dogliotti, E. The base excision repair: Mechanisms and its relevance for cancer susceptibility. Biochimie 2003, 85, 1053–1071. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Park, D.I.; Park, S.H.; Kim, S.H.; Kim, J.W.; Cho, Y.K.; Kim, H.J.; Sohn, C.I.; Jeon, W.K.; Kim, B.I.; Cho, E.Y.; et al. Effect of Helicobacter pylori infection on the expression of DNA mismatch repair protein. Helicobacter 2005, 10, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Tao, H.; Carloni, E.; Leung, W.K.; Graham, D.Y.; Sepulveda, A.R. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology 2002, 123, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.Z.; O’Hara, A.M.; Denning, T.L.; Dirden-Kramer, B.; Mifflin, R.C.; Reyes, V.E.; Ryan, K.A.; Elliott, S.N.; Izumi, T.; Boldogh, I.; et al. Helicobacter pylori and h2o2 increase ap endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology 2004, 127, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Den Hartog, G.; Chattopadhyay, R.; Ablack, A.; Hall, E.H.; Butcher, L.D.; Bhattacharyya, A.; Eckmann, L.; Harris, P.R.; Das, S.; Ernst, P.B.; et al. Regulation of Rac1 and reactive oxygen species production in response to infection of gastrointestinal epithelia. PLoS Pathog. 2016, 12, e1005382. [Google Scholar] [CrossRef] [PubMed]
- Teoule, R.; Bert, C.; Bonicel, A. Thymine fragment damage retained in the DNA polynucleotide chain after gamma irradiation in aerated solutions. II. Radiat. Res. 1977, 72, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Altieri, F.; Grillo, C.; Maceroni, M.; Chichiarelli, S. DNA damage and repair: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 891–937. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef] [PubMed]
- Touati, E.; Michel, V.; Thiberge, J.M.; Ave, P.; Huerre, M.; Bourgade, F.; Klungland, A.; Labigne, A. Deficiency in OGG1 protects against inflammation and mutagenic effects associated with H. pylori infection in mouse. Helicobacter 2006, 11, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, J.P.; Dolwani, S.; Sampson, J.R. Inherited defects in the DNA glycosylase MYH cause multiple colorectal adenoma and carcinoma. Carcinogenesis 2003, 24, 1281–1282; author reply 1283. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Tominaga, Y.; Yamauchi, K.; Nakatsu, Y.; Sakumi, K.; Yoshiyama, K.; Egashira, A.; Kura, S.; Yao, T.; Tsuneyoshi, M.; et al. Mutyh-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007, 67, 6599–6604. [Google Scholar] [CrossRef] [PubMed]
- Nossa, C.W.; Blanke, S.R. Helicobacter pylori activation of PARP-1: Usurping a versatile regulator of host cellular health. Gut Microbes 2010, 1, 373–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabley, J.G.; Pacher, P.; Deb, A.; Wallace, R.; Elder, R.H.; Szabo, C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005, 19, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Burnette, B.R.; Cross, J.V.; Mitra, S.; Ernst, P.B.; Bhakat, K.K.; Crowe, S.E. Acetylation of apurinic/apyrimidinic endonuclease-1 regulates Helicobacter pylori-mediated gastric epithelial cell apoptosis. Gastroenterology 2009, 136, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Bhattacharyya, A.; Mifflin, R.C.; Smith, M.F.; Ryan, K.A.; Scott, K.G.; Naganuma, M.; Casola, A.; Izumi, T.; Mitra, S.; et al. Interleukin-8 induction by helicobacter pylori in gastric epithelial cells is dependent on apurinic/apyrimidinic endonuclease-1/redox factor-1. J. Immunol. 2006, 177, 7990–7999. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Tang, J.T.; Peng, Y.S.; Chen, X.Y.; Zhang, Y.J.; Fang, J.Y. Xrcc1 downregulated through promoter hypermethylation is involved in human gastric carcinogenesis. J. Dig. Dis. 2010, 11, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Rashid, I.; Tomkinson, A.E.; Wallace, S.S.; Pederson, D.S. The human ligase iiialpha-xrcc1 protein complex performs DNA nick repair after transient unwrapping of nucleosomal DNA. J. Biol. Chem. 2017, 292, 5227–5238. [Google Scholar] [CrossRef] [PubMed]
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. 2015, 11, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Klungland, A.; Hoss, M.; Gunz, D.; Constantinou, A.; Clarkson, S.G.; Doetsch, P.W.; Bolton, P.H.; Wood, R.D.; Lindahl, T. Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell 1999, 3, 33–42. [Google Scholar] [CrossRef]
- Lee, H.S.; Choe, G.; Park, K.U.; Park, D.J.; Yang, H.K.; Lee, B.L.; Kim, W.H. Altered expression of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) during gastric carcinogenesis and its clinical implications on gastric cancer. Int. J. Oncol. 2007, 31, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Lim, J.W.; Kim, H. Oxidative DNA damage response in Helicobacter pylori-infected mongolian gerbils. J. Cancer Prev. 2013, 18, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.D.; Ferguson, D.O.; Alt, F.W. The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 2003, 194, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Yoshida, H.; Ogura, K.; Mitsuno, Y.; Hirata, Y.; Yamaji, Y.; Akanuma, M.; Shiratori, Y.; Omata, M. H. Pylori activates NF-κB through a signaling pathway involving IκB kinases, NF-κB -inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology 2000, 119, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Pathak, S.; Kundu, M.; Basu, J. Mitogen-activated protein kinases regulate mycobacterium avium-induced tumor necrosis factor-α release from macrophages. FEMS Immunol. Med. Microbiol. 2002, 34, 73–80. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Iaquinto, G. Helicobacter pylori and gastric diseases: A dangerous association. Cancer Lett. 2004, 213, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.; Chen, L.F. The many roads traveled by Helicobacter pylori to NF-κB activation. Gut Microbes 2010, 1, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Baldwin, A.S., Jr. NF-κB as a therapeutic target in cancer. Trends Mol. Med. 2002, 8, 385–389. [Google Scholar] [CrossRef]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Oussaief, L.; Ramirez, V.; Hippocrate, A.; Arbach, H.; Cochet, C.; Proust, A.; Raphael, M.; Khelifa, R.; Joab, I. NF-κB-mediated modulation of inducible nitric oxide synthase activity controls induction of the epstein-barr virus productive cycle by transforming growth factor β1. J. Virol. 2011, 85, 6502–6512. [Google Scholar] [CrossRef] [PubMed]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Memet, S.; Huerre, M.R.; Coyle, A.J.; et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Kwok, T.; Hartig, R.; Konig, W.; Backert, S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori caga protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef] [PubMed]
- Geem, D.; Medina-Contreras, O.; Kim, W.; Huang, C.S.; Denning, T.L. Isolation and characterization of dendritic cells and macrophages from the mouse intestine. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.A.; Di Silvio, M.; Nussler, A.K.; Wang, S.C.; Shapiro, R.A.; Simmons, R.L.; Billiar, T.R. Nitric oxide synthase expression is induced in hepatocytes in vivo during hepatic inflammation. J. Surg. Res. 1993, 55, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Nussler, A.K.; Geller, D.A.; Sweetland, M.A.; Di Silvio, M.; Billiar, T.R.; Madariaga, J.B.; Simmons, R.L.; Lancaster, J.R., Jr. Induction of nitric oxide synthesis and its reactions in cultured human and rat hepatocytes stimulated with cytokines plus LPS. Biochem. Biophys. Res. Commun. 1993, 194, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, C.J.; Padalko, E. iNOS (NOS2) at a glance. J. Cell Sci. 2004, 117, 2865–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, L.L.; Lawton, F.G.; Knowles, R.G.; Beesley, J.E.; Riveros-Moreno, V.; Moncada, S. Nitric oxide synthase activity in human gynecological cancer. Cancer Res. 1994, 54, 1352–1354. [Google Scholar] [PubMed]
- Rieder, G.; Hofmann, J.A.; Hatz, R.A.; Stolte, M.; Enders, G.A. Up-regulation of inducible nitric oxide synthase in Helicobacter pylori-associated gastritis may represent an increased risk factor to develop gastric carcinoma of the intestinal type. Int. J. Med. Microbiol. 2003, 293, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Touati, E.; Michel, V.; Thiberge, J.M.; Wuscher, N.; Huerre, M.; Labigne, A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology 2003, 124, 1408–1419. [Google Scholar] [CrossRef]
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; Allen, J.S.; et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 1991, 254, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, H.H.; Cherian, M.A.; Green, P.; Ratner, L. Inducible nitric oxide synthase mediates DNA double strand breaks in human T-cell leukemia virus type 1-induced leukemia/lymphoma. Retrovirology 2015, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Brunson, D.; Crespi, C.L.; Penman, B.W.; Wishnok, J.S.; Tannenbaum, S.R. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc. Natl. Acad. Sci. USA 1992, 89, 3030–3034. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.B.; Libby, P.; Liao, J.K. Induction and stabilization of IκBα by nitric oxide mediates inhibition of NF-κB. J. Biol. Chem. 1995, 270, 14214–14219. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.R.; Botting, C.H.; Panico, M.; Morris, H.R.; Hay, R.T. Inhibition of NF-κB DNA binding by nitric oxide. Nucleic Acids Res. 1996, 24, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.A.; Punzalan, C.; Kuo, P.C. Endotoxin-mediated S-nitrosylation of p50 alters NF-κB -dependent gene transcription in ANA-1 murine macrophages. J. Immunol. 1999, 162, 4101–4108. [Google Scholar]
- Park, J.M.; Greten, F.R.; Wong, A.; Westrick, R.J.; Arthur, J.S.; Otsu, K.; Hoffmann, A.; Montminy, M.; Karin, M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis—CREB and NF-κB as key regulators. Immunity 2005, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The enzymatic and DNA binding activity of PARP-1 are not required for NF-κB coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, O.; Diestel, A.; Eyupoglu, I.Y.; Nitsch, R. Regulation of microglial expression of integrins by poly (ADP-ribose) polymerase-1. Nat. Cell Biol. 2001, 3, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hao, L.; Wang, Y.; Qin, W.; Wang, X.; Zhao, T.; Liu, Y.; Sheng, L.; Du, Y.; Zhang, M.; et al. Inhibition of poly (ADP-ribose) polymerase and inducible nitric oxide synthase protects against ischemic myocardial damage by reduction of apoptosis. Mol. Med. Rep. 2015, 11, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H.; Zhu, L.; Beckman, J.S. Peroxynitrite formation from macrophage-derived nitric oxide. Arch. Biochem. Biophys. 1992, 298, 446–451. [Google Scholar] [CrossRef]
- Kong, S.K.; Yim, M.B.; Stadtman, E.R.; Chock, P.B. Peroxynitrite disables the tyrosine phosphorylation regulatory mechanism: Lymphocyte-specific tyrosine kinase fails to phosphorylate nitrated cdc2(6-20)NH2 peptide. Proc. Natl. Acad. Sci. USA 1996, 93, 3377–3382. [Google Scholar] [CrossRef] [PubMed]
- Starke, D.W.; Chen, Y.; Bapna, C.P.; Lesnefsky, E.J.; Mieyal, J.J. Sensitivity of protein sulfhydryl repair enzymes to oxidative stress. Free Radic. Biol. Med. 1997, 23, 373–384. [Google Scholar] [CrossRef]
- Wink, D.A.; Laval, J. The Fpg protein, a DNA repair enzyme, is inhibited by the biomediator nitric oxide in vitro and in vivo. Carcinogenesis 1994, 15, 2125–2129. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.R.; Graves, R.J.; de Murcia, G.; Castaing, B.; Laval, J. Fpg protein of escherichia coli is a zinc finger protein whose cysteine residues have a structural and/or functional role. J. Biol. Chem. 1993, 268, 9063–9070. [Google Scholar] [PubMed]
- Telford, J.L.; Covacci, A.; Rappuoli, R.; Chiara, P. Immunobiology of helicobacter pylori infection. Curr. Opin. Immunol. 1997, 9, 498–503. [Google Scholar] [CrossRef]
- Le May, N.; Fradin, D.; Iltis, I.; Bougneres, P.; Egly, J.M. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol. Cell 2012, 47, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Mota-Fernandes, D.; Velez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Leibeling, D.; Laspe, P.; Emmert, S. Nucleotide excision repair and cancer. J. Mol. Histol. 2006, 37, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. How nucleotide excision repair protects against cancer. Nat. Rev. Cancer 2001, 1, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Liu, J.W.; Sun, L.P.; Xu, Q.; Duan, Z.P.; Dong, N.N.; Yuan, Y. Expression of XPG protein in the development, progression and prognosis of gastric cancer. PLoS ONE 2014, 9, e108704. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, E.L.; Haber, J.E. DNA repair: Rad alert. Curr. Biol. 1997, 7, R492–R495. [Google Scholar] [CrossRef]
- Kanaar, R.; Hoeijmakers, J.H. Recombination and joining: Different means to the same ends. Genes Funct. 1997, 1, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Tauchi, H.; Nakamura, A.; Kondo, N.; Sakamoto, S.; Endo, S.; Smeets, D.; Solder, B.; Belohradsky, B.H.; Der Kaloustian, V.M.; et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat. Genet. 1998, 19, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R., 3rd; Hays, L.; Morgan, W.F.; Petrini, J.H. The hmre11/hrad50 protein complex and nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef]
- Stracker, T.H.; Petrini, J.H. The mre11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Lehmann, A.R. Maintaining integrity. Nat. Cell Biol. 2004, 6, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. Phosphatases join kinases in DNA-damage response pathways. Trends Cell Biol. 2004, 14, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Dupre, A.; Boyer-Chatenet, L.; Gautier, J. Two-step activation of ATM by DNA and the Mre11–Rad50–Nbs1 complex. Nat. Struct. Mol. Biol. 2006, 13, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Banin, S.; Ouyang, H.; Li, G.C.; Courtois, G.; Shiloh, Y.; Karin, M.; Rotman, G. ATM is required for IκB kinase (IKKk) activation in response to DNA double strand breaks. J. Biol. Chem. 2001, 276, 8898–8903. [Google Scholar] [CrossRef] [PubMed]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Ishiai, M.; Hosono, Y.; Yoshimura, A.; Tada, S.; Adachi, N.; Koyama, H.; Takata, M.; Takeda, S.; Enomoto, T.; et al. Ku70/80, DNA-PKcs, and artemis are essential for the rapid induction of apoptosis after massive DSB formation. Cell. Signal. 2008, 20, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.W.; Kim, H.; Kim, K.H. Expression of Ku70 and Ku80 mediated by NF-κB and cyclooxygenase-2 is related to proliferation of human gastric cancer cells. J. Biol. Chem. 2002, 277, 46093–46100. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Y.; Lim, J.W.; Kim, H.; Morio, T.; Kim, K.H. Oxidative stress induces nuclear loss of DNA repair proteins Ku70 and Ku80 and apoptosis in pancreatic acinar AR42J cells. J. Biol. Chem. 2003, 278, 36676–36687. [Google Scholar] [CrossRef] [PubMed]
- Volcic, M.; Karl, S.; Baumann, B.; Salles, D.; Daniel, P.; Fulda, S.; Wiesmuller, L. NF-κB regulates DNA double-strand break repair in conjunction with BRCA1–CtIP complexes. Nucleic Acids Res. 2012, 40, 181–195. [Google Scholar] [CrossRef] [PubMed]


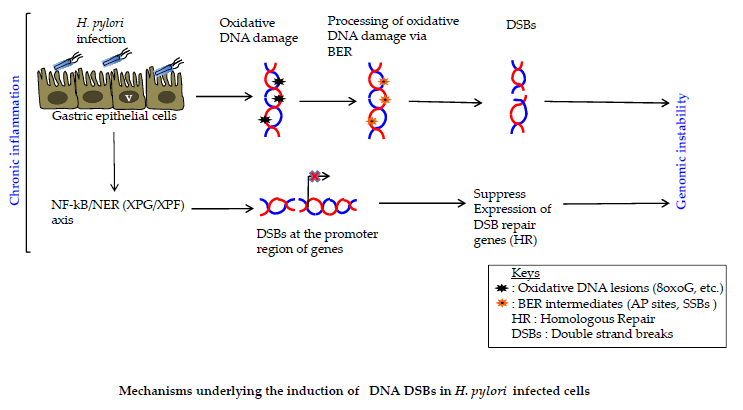{kind=link}
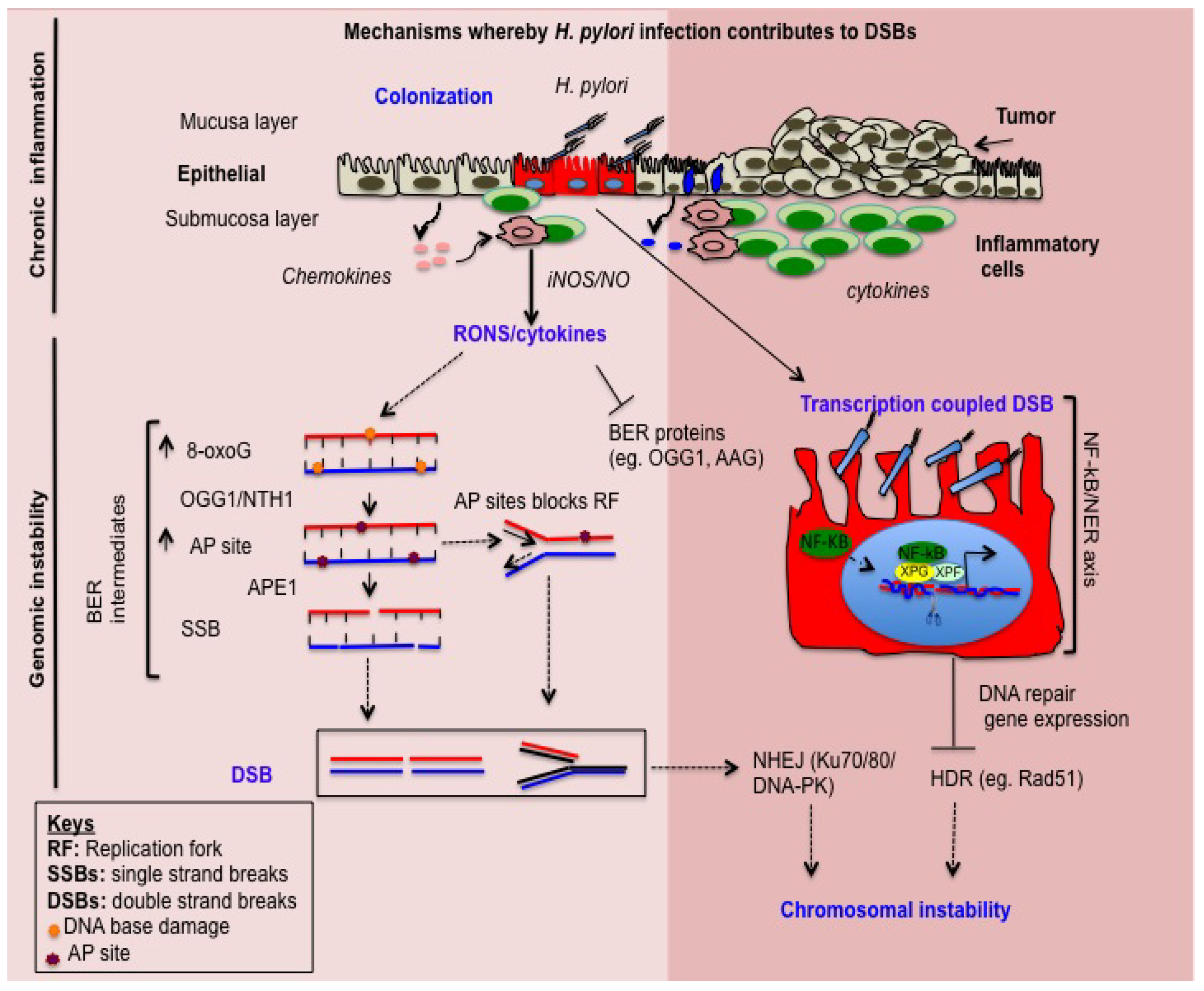{kind=link}
| Gene | Role of Gene Products | Interplay between H. pylori & Gene | References |
|---|---|---|---|
| BER | |||
| OGG1 | removes 8oxoG and FapyG DNA lesions | absence causes increased mutation frequency, fewer DSBs and decreased inflammation | [26,55,61] |
| NEIL1 | removes 8oxoG and Tg lesions | decreases mRNA in tumor; unknown role during infection | [34] |
| APE1 | acts as a negative regulator of ROS and enhances chemokine release | enhances the expression of mRNA and protein | [49,62,63] |
| POLB | removes 5′-dRP group and adds a single nucleotide base | infection does not affect gene expression and protein level | [26] |
| XRCC1 | scaffold protein enhance ligation | downregulated via promoter hypermethylation | [64,65] |
| NER | |||
| XPG | cuts the 3′ of the DNA damage site; forms complex with NF-κB and promotes target gene expression | moderates change in gene expression | [11,66,67] |
| XPF | forms complex with NF-κB & promotes targeted gene expression | moderates change in gene expression | [11,66] |
| XPA | recognition bulk DNA adduct | increases IL-8 cytokine expression | [11,66] |
| NHEJ | |||
| DNA-PK | increases cellular proliferation & facilitates NHEJ (nonhomologous DNA end-joining) repair | enhances activity and expression | [68] |
| Ku70/80 | protects DNA DSB ends and prevents cell death | decreases protein level | [69] |
| DNA ligase IV | completes NHEJ repair by sealing DNA DSB regions | knock-down enhances DSBs | [11] |
| XRCC4 | scaffold to hold DNA DSBs ends to enhance ligation | knock-down promotes DNA DSBs | [11] |
| HR | |||
| NBS1 | DNA DSB end processing/DDR | decreases expression and may impair DNA end processing and DDR | [66] |
| Rad51 | strand exchange and enhances DSB repair | decreases gene expression; however, infection does not increase DSBs | [25] |
| RPA1 | ssDNA binding and DDR | downregulates mRNA | [66] |
| Mre11 | DSB end processing and DDR | decreases expression and impairs end processing and DDR | [66] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kidane, D. Molecular Mechanisms of H. pylori-Induced DNA Double-Strand Breaks. Int. J. Mol. Sci. 2018, 19, 2891. https://doi.org/10.3390/ijms19102891
Kidane D. Molecular Mechanisms of H. pylori-Induced DNA Double-Strand Breaks. International Journal of Molecular Sciences. 2018; 19(10):2891. https://doi.org/10.3390/ijms19102891
Chicago/Turabian StyleKidane, Dawit. 2018. "Molecular Mechanisms of H. pylori-Induced DNA Double-Strand Breaks" International Journal of Molecular Sciences 19, no. 10: 2891. https://doi.org/10.3390/ijms19102891
APA StyleKidane, D. (2018). Molecular Mechanisms of H. pylori-Induced DNA Double-Strand Breaks. International Journal of Molecular Sciences, 19(10), 2891. https://doi.org/10.3390/ijms19102891