Conformational Dynamics of Herpesviral NEC Proteins in Different Oligomerization States


Abstract
:
1. Introduction
2. Results and Discussion
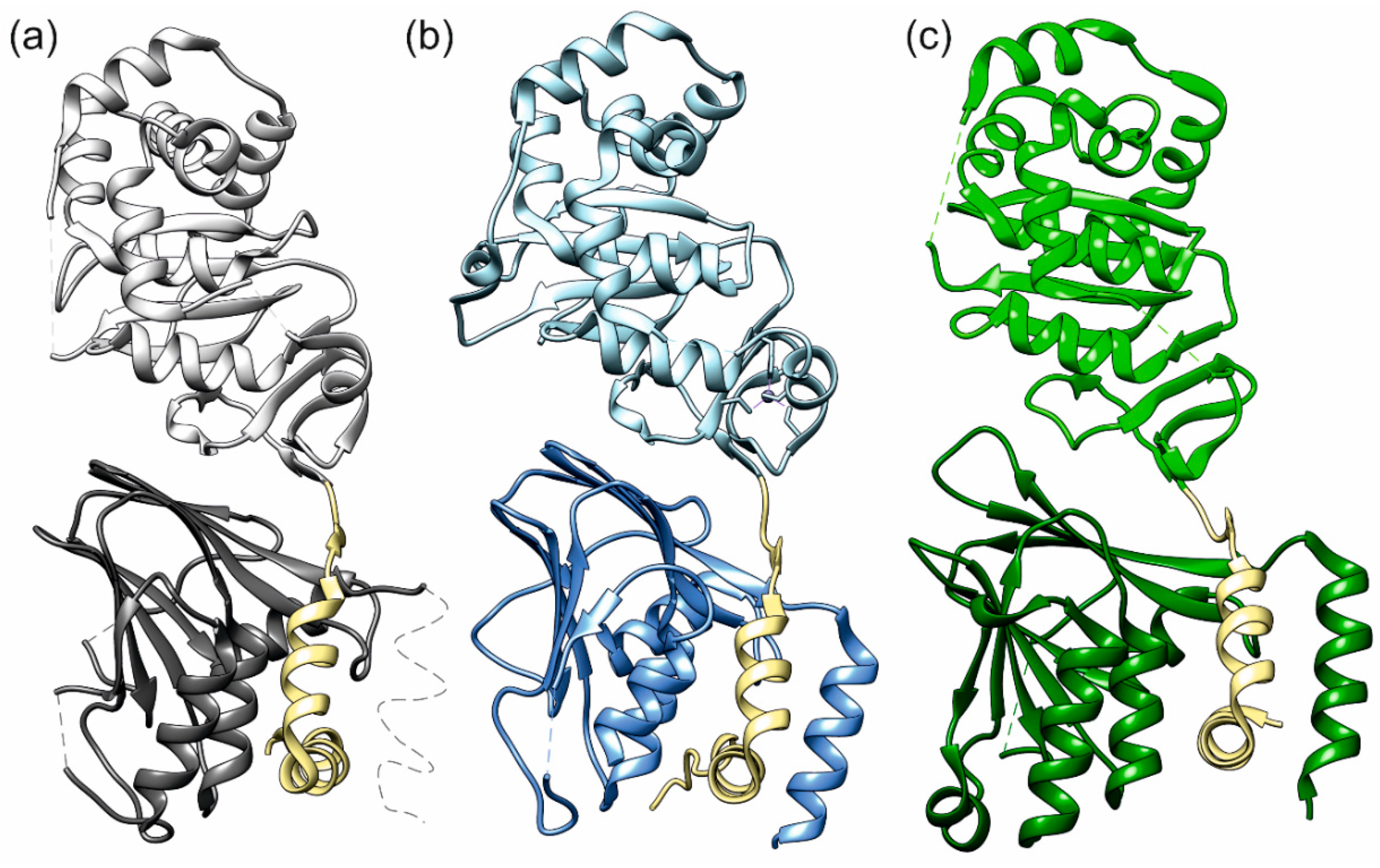
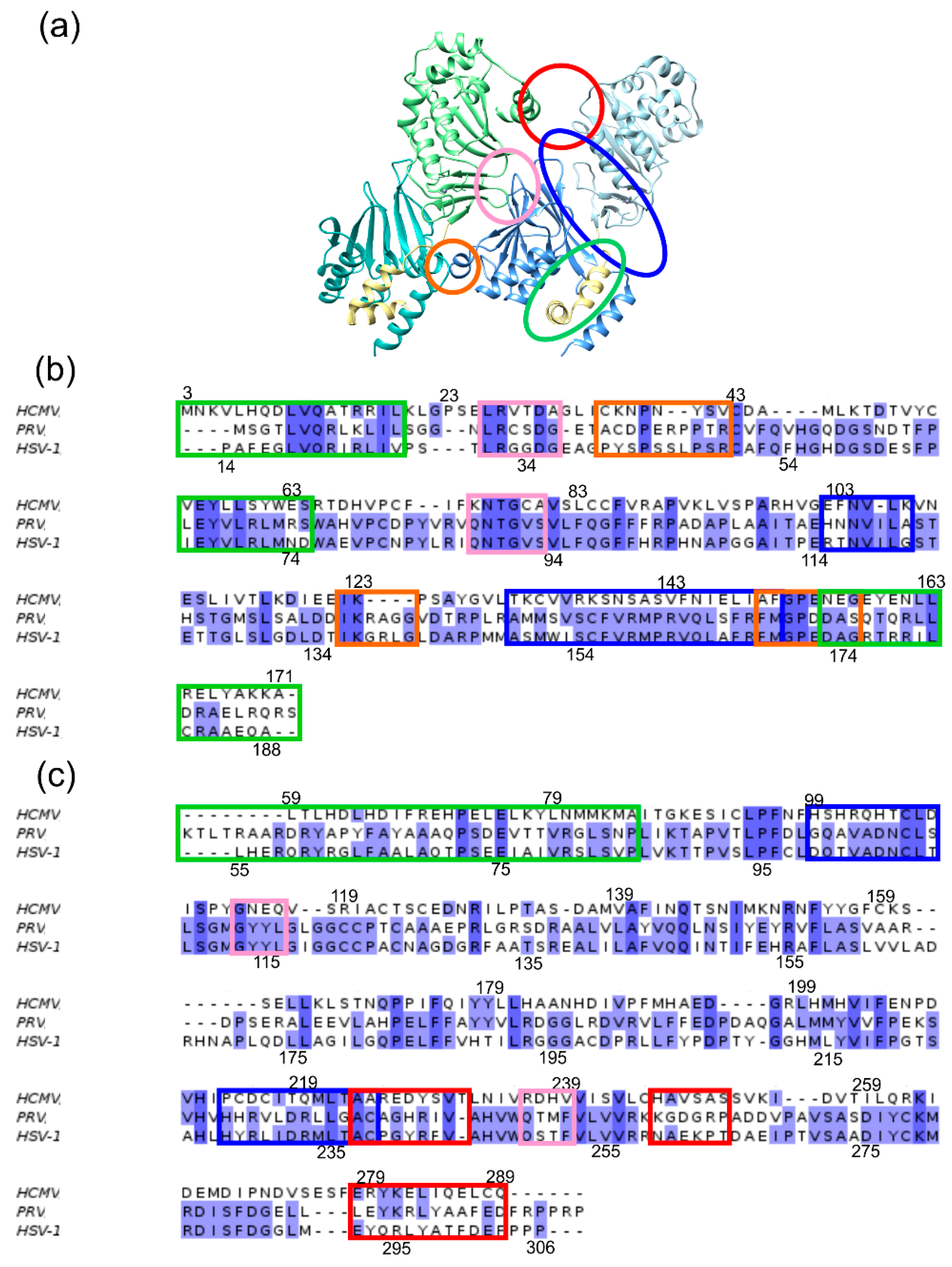
2.1. Interface Analysis of the NECs (Nuclear Egress Complexes)
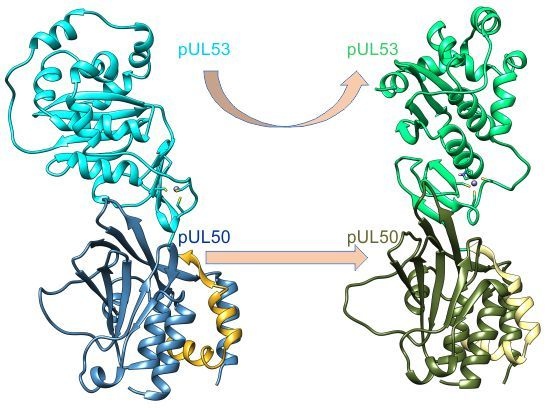
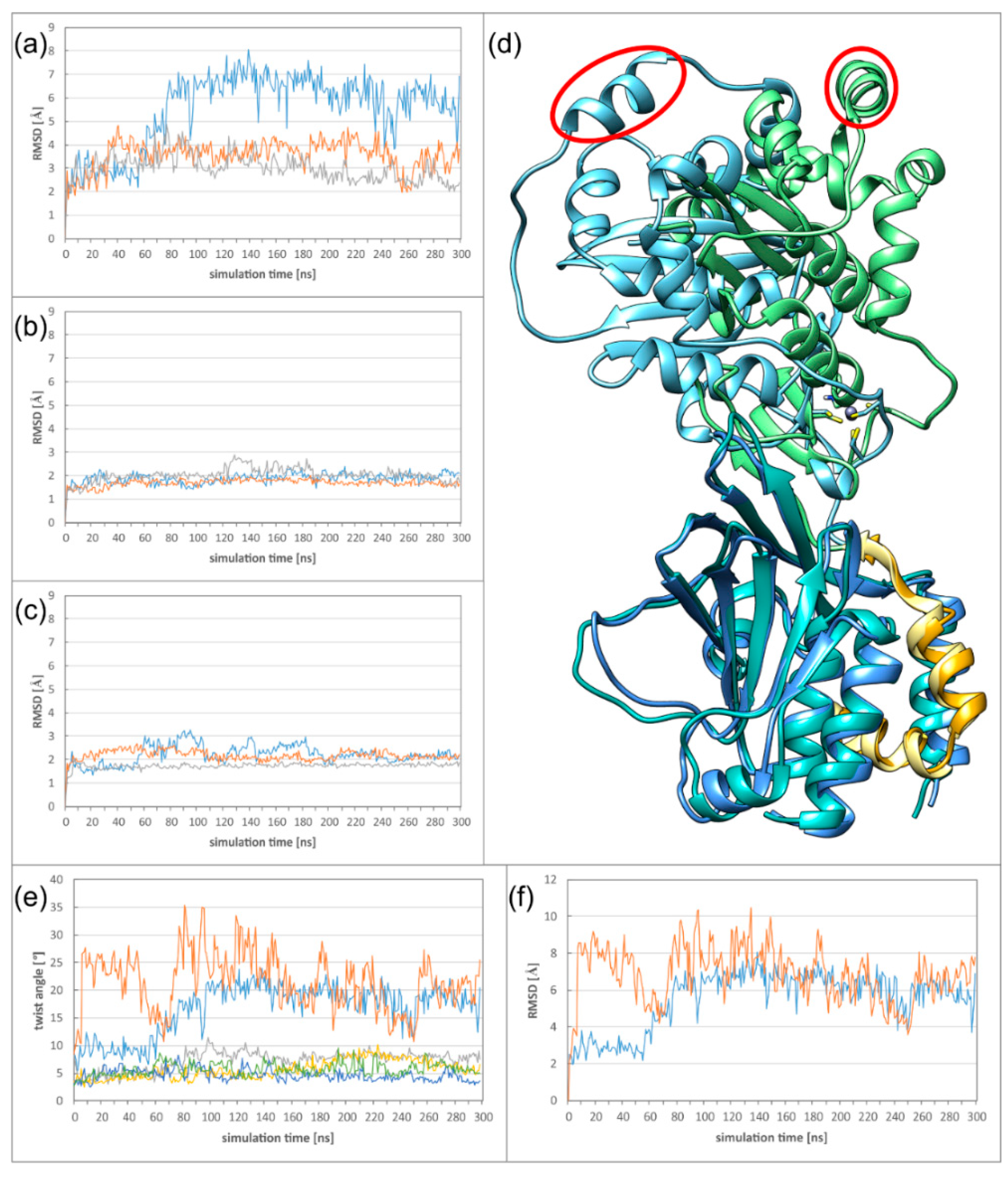
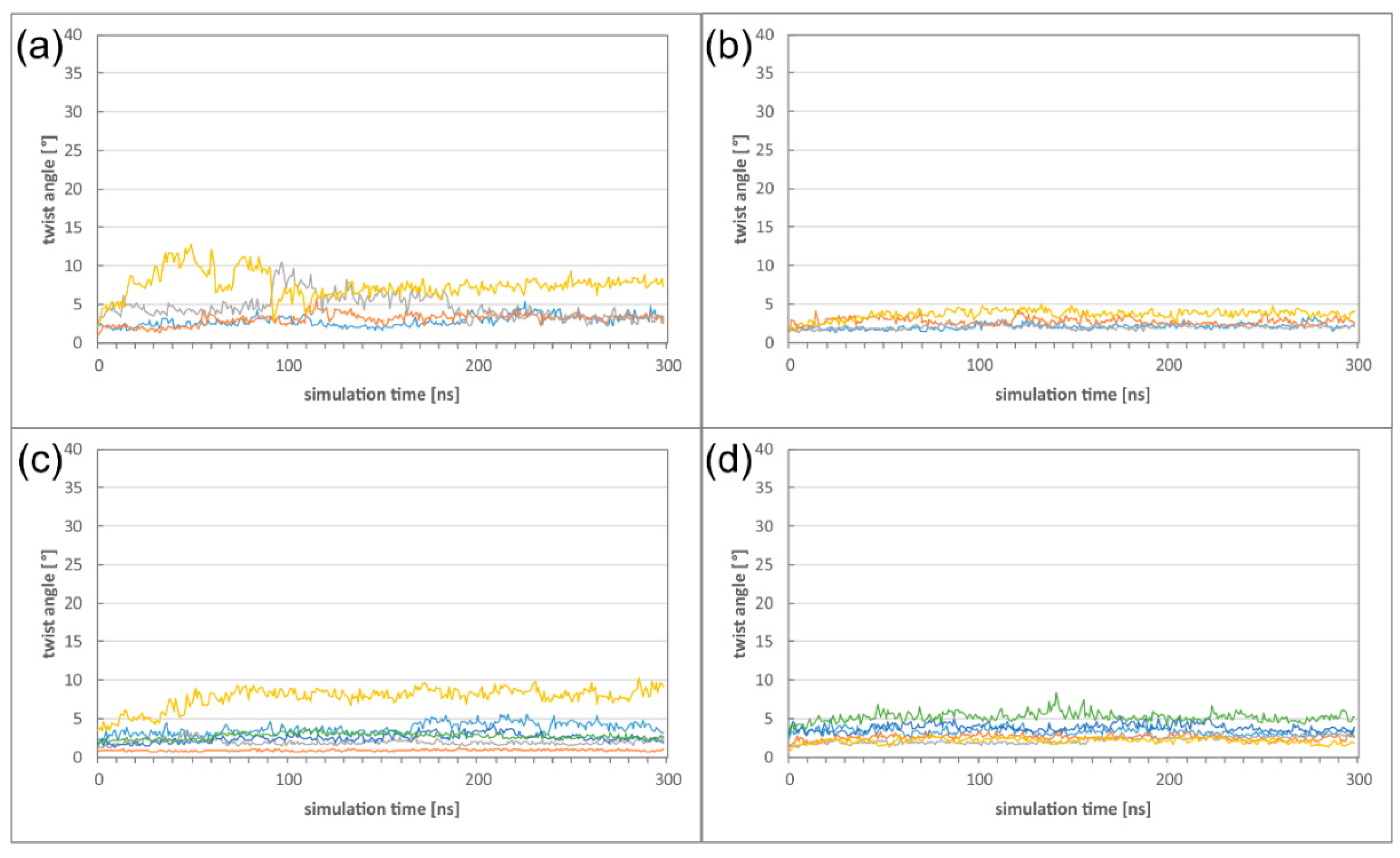
2.2. The HCMV NEC Heterodimer Is Considerably More Flexible Than Its Counterparts in PRV and HSV-1
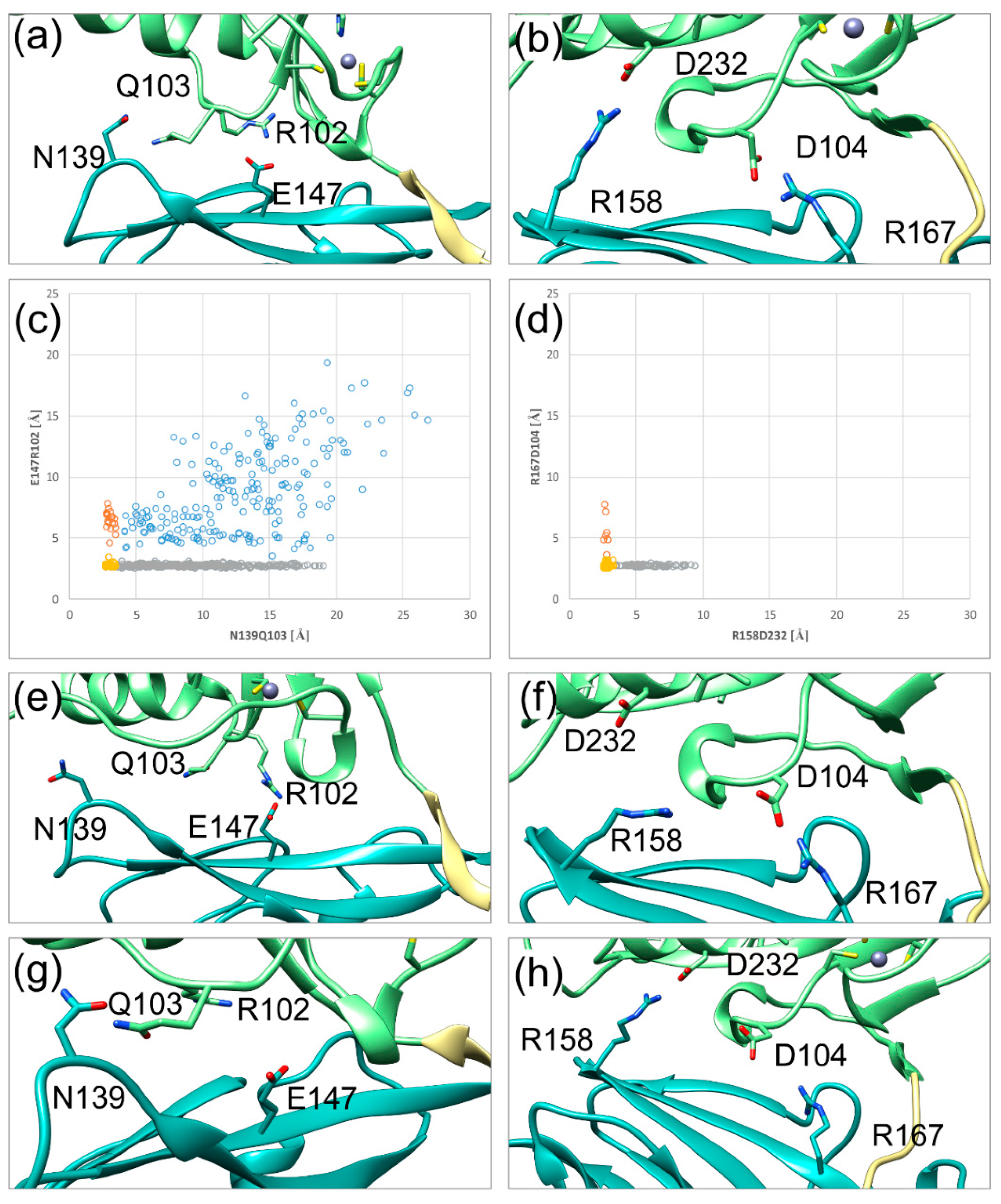
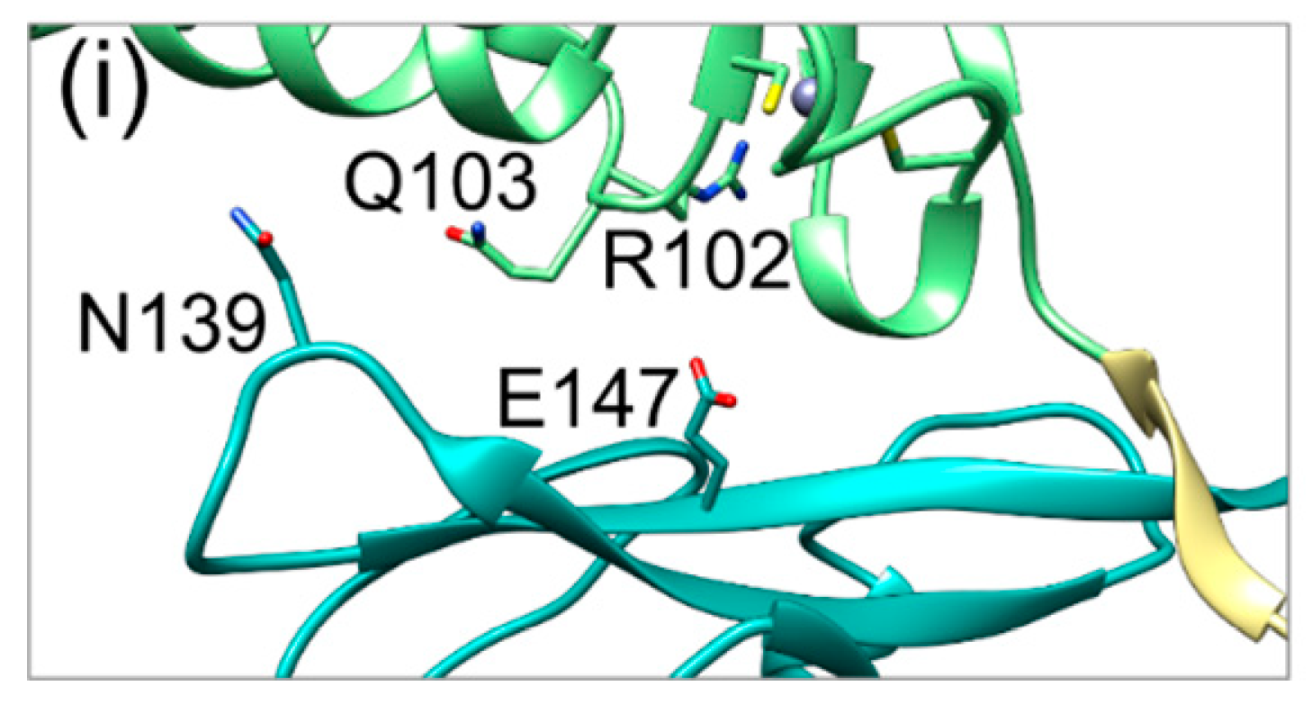
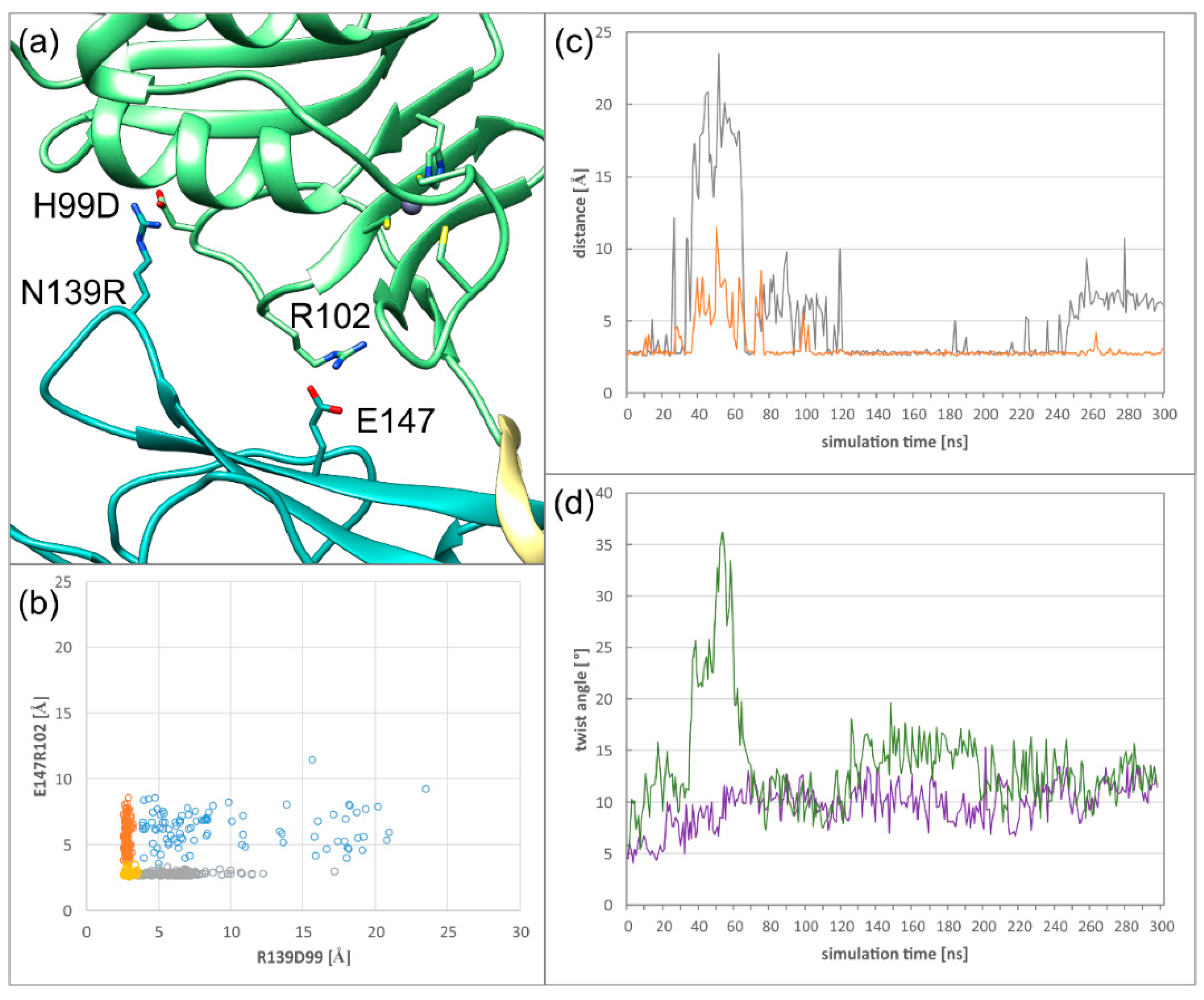
2.3. The Secondary Interface Is a Critical Determinant of Subdomain Orientation
2.4. HCMV NEC Secondary Interface Can Be Stabilized by Introducing an Extra Salt bridge
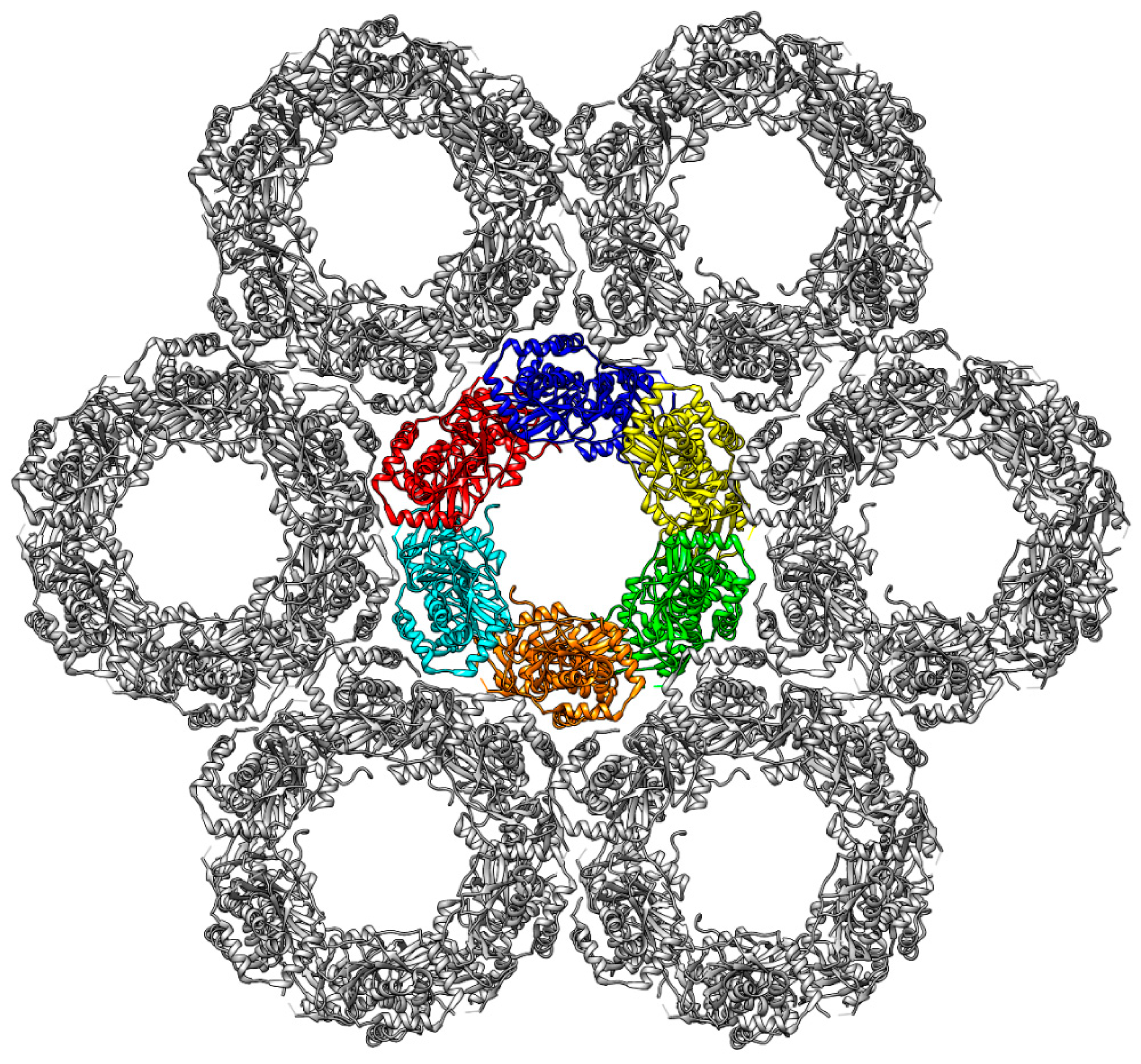
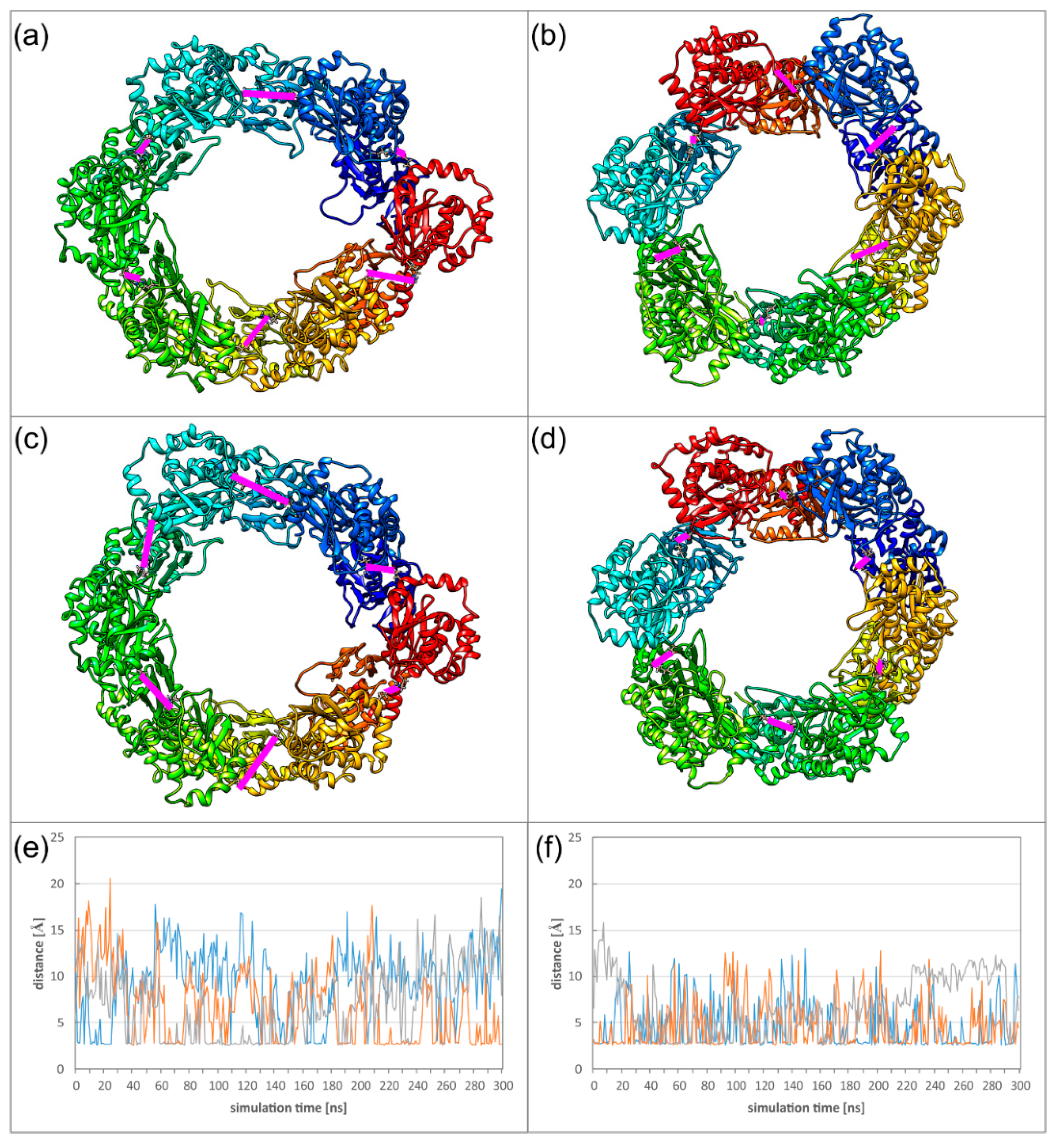
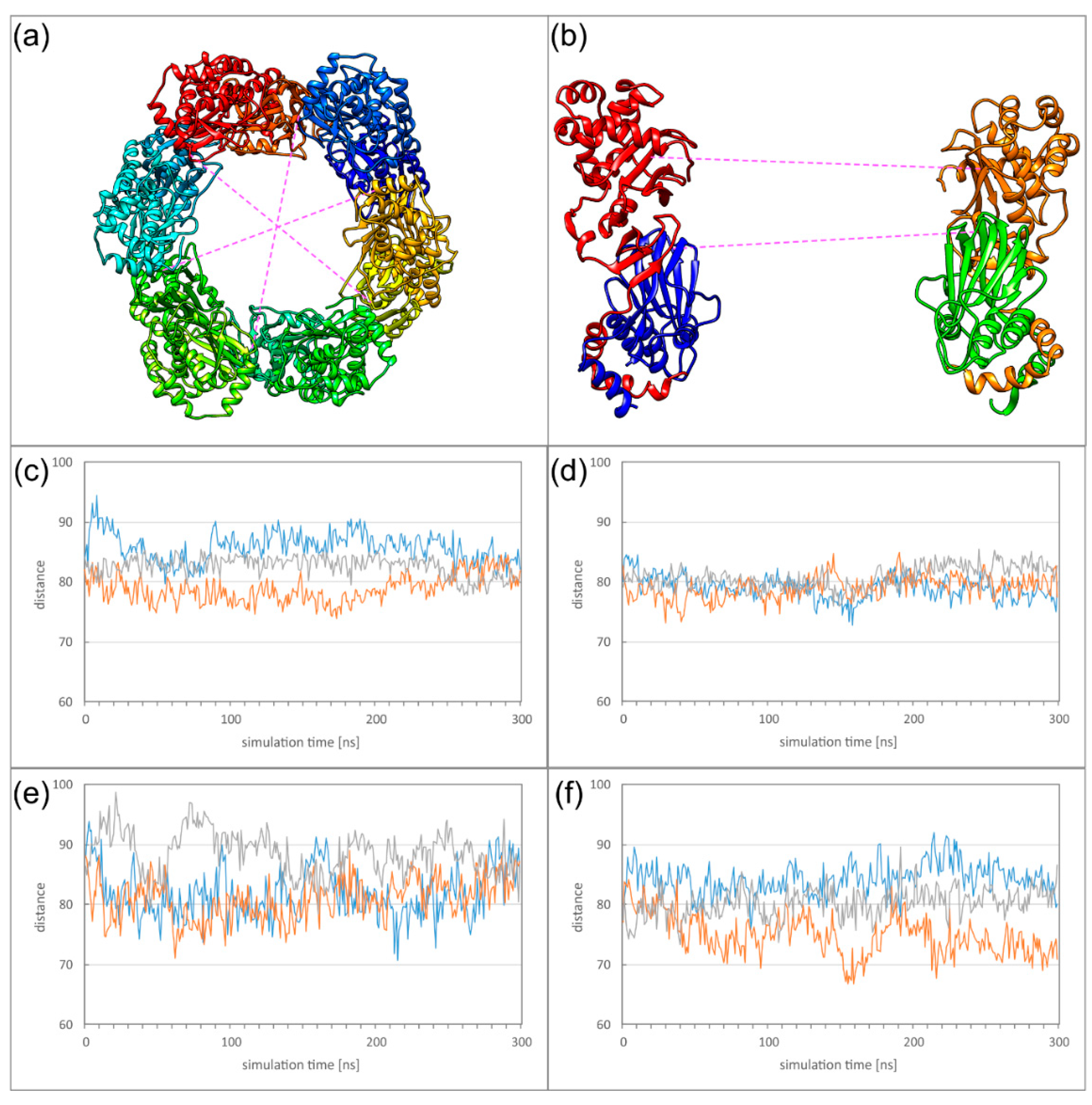
2.5. Dyamics of Higher NEC Oligomers
3. Methods
3.1. Processing of X-ray Structures
3.2. MD Setup
3.3. Twist Angle Calculation
3.4. Other Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | molecular dynamics |
| HSV-1 | herpes simplex virus 1 |
| PRV | pseudorabies virus |
| HCMV | human cytomegalovirus |
| INM | inner nuclear membrane |
| ONM | outer nuclear membrane |
| ER | endoplasmic reticulum |
| cryo-EM | cryo-electron microscopy |
| NEC | nuclear egress complex |
| PIF | primary interface |
| SIF | secondary interface |
| OIF | oligomer interface |
| MCP | major capsid protein |
| RMSD | root-mean-square deviation |
| RMSF | root-mean-square fluctuation |
| PDB | protein data bank |
References
- Andrés, G.; García-Escudero, R.; Simón-Mateo, C.; Viñuela, E. African swine fever virus is enveloped by a two-membraned collapsed cisterna derived from the endoplasmic reticulum. J. Virol. 1998, 72, 8988–9001. [Google Scholar] [PubMed]
- Hiller, G.; Weber, K. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 1985, 55, 651–659. [Google Scholar] [PubMed]
- McMillan, T.N.; Johnson, D.C. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 2001, 75, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C. Budding events in herpesvirus morphogenesis. Virus Res. 2004, 106, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Sternberg, M.R.; Kottiri, B.J.; McQuillan, G.M.; Lee, F.K.; Nahmias, A.J.; Berman, S.M.; Markowitz, L.E. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 2006, 296, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Fowler, K.B.; McCollister, F.P.; Dahle, A.J.; Boppana, S.; Britt, W.J.; Pass, R.F. Progressive and fluctuating sensorineural hearing loss in children with asymptomatic congenital cytomegalovirus infection. J. Pediatr. 1997, 130, 624–630. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef]
- Hollinshead, M.; Johns, H.L.; Sayers, C.L.; Gonzalez-Lopez, C.; Smith, G.L.; Elliott, G. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 2012, 31, 4204–4220. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed]
- Sam, M.D.; Evans, B.T.; Coen, D.M.; Hogle, J.M. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J. Virol. 2009, 83, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Feichtinger, S.; Milbradt, J. Regulatory roles of protein kinases in cytomegalovirus replication. Adv. Virus Res. 2011, 80, 69–101. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Haas, J.; Wagner, M.; Krohne, G.; Koszinowski, U.H. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 2002, 297, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.; Guttmann, P.; Klupp, B.; Werner, S.; Rehbein, S.; Mettenleiter, T.C.; Schneider, G.; Grunewald, K. Correlative VIS-fluorescence and soft X-ray cryo-microscopy/tomography of adherent cells. J. Struct. Biol. 2012, 177, 193–201. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heuser, T.; Nicastro, D.; Heldwein, E.E. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat. Commun. 2014, 5, 4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roller, R.J.; Zhou, Y.; Schnetzer, R.; Ferguson, J.; DeSalvo, D. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 2000, 74, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Osterrieder, N.; Mettenleiter, T.C. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 2002, 76, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, J.M.; Heldwein, E.E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, K.E.; Sharma, M.; Mansueto, M.S.; Boeszoermenyi, A.; Filman, D.J.; Hogle, J.M.; Wagner, G.; Coen, D.M.; Arthanari, H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9010–9015. [Google Scholar] [CrossRef] [PubMed]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.J.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, C.; Dent, K.C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse, J.B.; Whittle, C.; Klupp, B.G.; Siebert, C.A.; Vasishtan, D.; Bäuerlein, F.J.B.; et al. Structural basis of vesicle formation at the inner nuclear membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Muller, Y.A.; Diewald, B.; Sticht, H.; Milbradt, J. The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 2017, 27. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, J.M.; Heldwein, E.E. Have NEC coat, will travel: Structural basis of membrane budding during nuclear egress in herpesviruses. Adv. Virus Res. 2017, 97, 107–141. [Google Scholar] [CrossRef] [PubMed]
- Zeev-Ben-Mordehai, T.; Weberruss, M.; Lorenz, M.; Cheleski, J.; Hellberg, T.; Whittle, C.; El Omari, K.; Vasishtan, D.; Dent, K.C.; Harlos, K.; et al. Crystal structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep. 2015, 13, 2645–2652. [Google Scholar] [CrossRef]
- Bjerke, S.L.; Cowan, J.M.; Kerr, J.K.; Reynolds, A.E.; Baines, J.D.; Roller, R.J. Effects of Charged Cluster Mutations on the Function of Herpes Simplex Virus Type 1 UL34 Protein. J. Virol. 2003, 77, 7601–7610. [Google Scholar] [CrossRef] [Green Version]
- Kurowski, M.A.; Bujnicki, J.M. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 2003, 31, 3305–3307. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2014, 47. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, T.E.; Miller, J.L.; Fox, T.; Darden, T.A.; Kollman, P.A. Molecular dynamics simulations on solvated biomolecular systems: The particle mesh ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An N⋅log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Wartha, F.; Horn, A.H.C.; Meiselbach, H.; Sticht, H. Molecular dynamics simulations of HIV-1 protease suggest different mechanisms contributing to drug resistance. J. Chem. Theory Comput. 2005, 1, 315–324. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Kim, S.; Voter, A.F.; Roitberg, A.E. Bad seeds sprout perilous dynamics: Stochastic thermostat induced trajectory synchronization in biomolecules. J. Chem. Theory Comput. 2009, 5, 1624–1631. [Google Scholar] [CrossRef]
- Uberuaga, B.P.; Anghel, M.; Voter, A.F. Synchronization of trajectories in canonical molecular-dynamics simulations: Observation, explanation, and exploitation. J. Chem. Phys. 2004, 120, 6363–6374. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Krosky, P.M.; Baek, M.-C.; Coen, D.M. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 2003, 77, 905–914. [Google Scholar] [CrossRef]
- Kuny, C.V.; Chinchilla, K.; Culbertson, M.R.; Kalejta, R.F. Cyclin-dependent kinase-like function is shared by the beta- and gamma- subset of the conserved herpesvirus protein kinases. PLoS Pathog. 2010, 6, e1001092. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Vollmer, B.; Unsay, J.D.; Klupp, B.G.; García-Sáez, A.J.; Mettenleiter, T.C.; Antonin, W. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J. Biol. Chem. 2015, 290, 6962–6974. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jih, J.; Jiang, J.; Zhou, Z.H. Atomic structure of the human cytomegalovirus capsid with its securing tegument layer of pp150. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Rönfeldt, S.; Klupp, B.G.; Franzke, K.; Mettenleiter, T.C. Lysine 242 within helix 10 of the pseudorabies virus nuclear egress complex pUL31 component is critical for primary envelopment of nucleocapsids. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, R.; Friedler, A. Allosteric modulation of protein oligomerization: An emerging approach to drug design. Front. Chem. 2014, 2, 9. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface | Inter-Action | HCMV | Inter-Action | HSV-1 | ||
|---|---|---|---|---|---|---|
| DimDim | Hex | DimDim | Hex | |||
| OIF I | E282R225 | 16 | 14 | E302K262 | 35 | 30 |
| OIF II | E115R27 | 64 | 77 | S250G91 | 67 | 46 |
| OIF III | E156K36 | 53 | 58 | R139S48 | 61 | 43 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diewald, B.; Socher, E.; Söldner, C.A.; Sticht, H. Conformational Dynamics of Herpesviral NEC Proteins in Different Oligomerization States. Int. J. Mol. Sci. 2018, 19, 2908. https://doi.org/10.3390/ijms19102908
Diewald B, Socher E, Söldner CA, Sticht H. Conformational Dynamics of Herpesviral NEC Proteins in Different Oligomerization States. International Journal of Molecular Sciences. 2018; 19(10):2908. https://doi.org/10.3390/ijms19102908
Chicago/Turabian StyleDiewald, Benedikt, Eileen Socher, Christian A. Söldner, and Heinrich Sticht. 2018. "Conformational Dynamics of Herpesviral NEC Proteins in Different Oligomerization States" International Journal of Molecular Sciences 19, no. 10: 2908. https://doi.org/10.3390/ijms19102908
APA StyleDiewald, B., Socher, E., Söldner, C. A., & Sticht, H. (2018). Conformational Dynamics of Herpesviral NEC Proteins in Different Oligomerization States. International Journal of Molecular Sciences, 19(10), 2908. https://doi.org/10.3390/ijms19102908