The Main Determinants of Diabetes Mellitus Vascular Complications: Endothelial Dysfunction and Platelet Hyperaggregation


 ,
,  and
and
Abstract
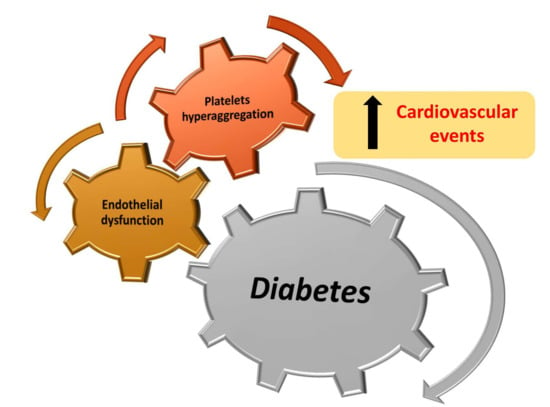
:
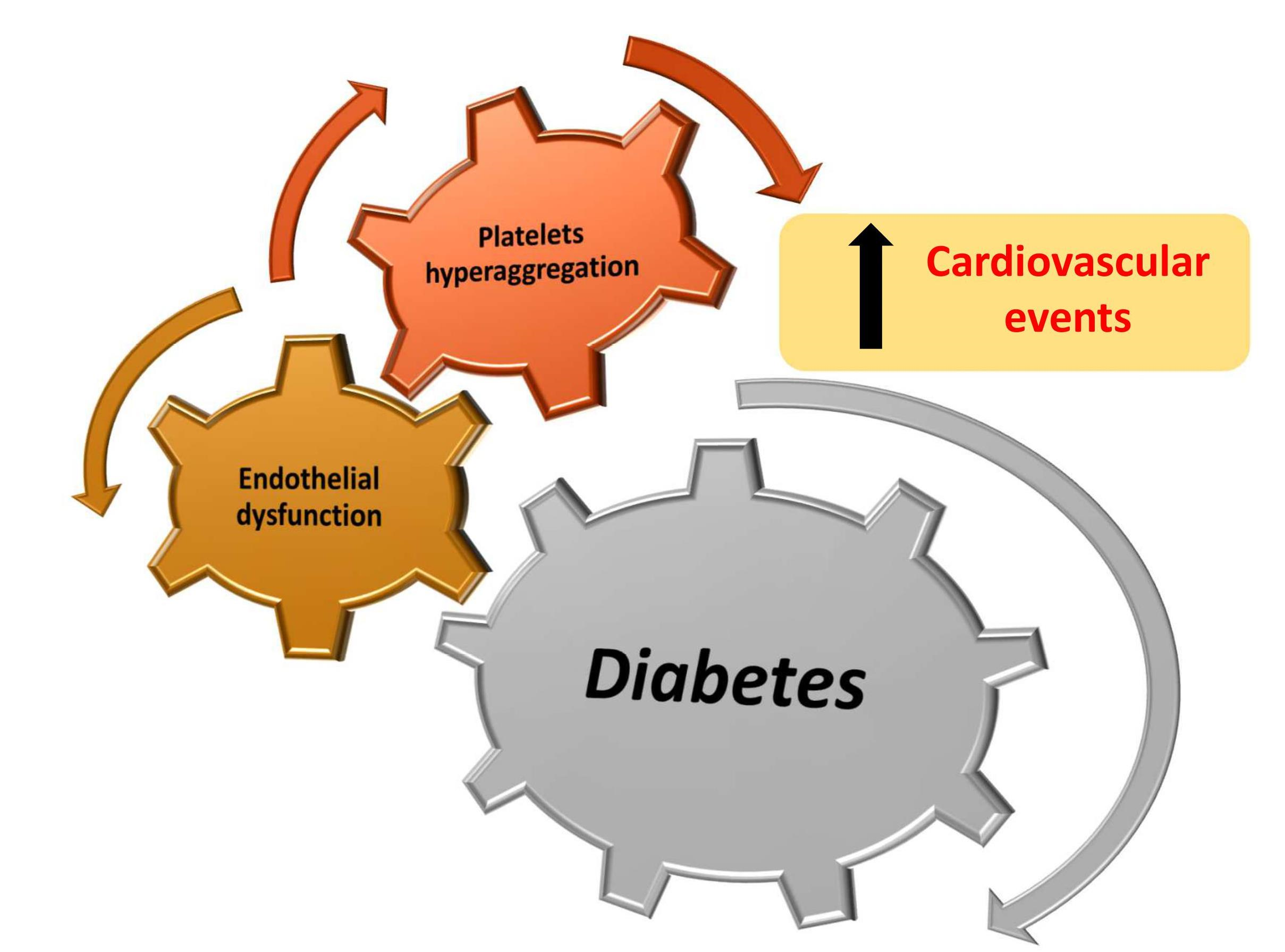{kind=link}
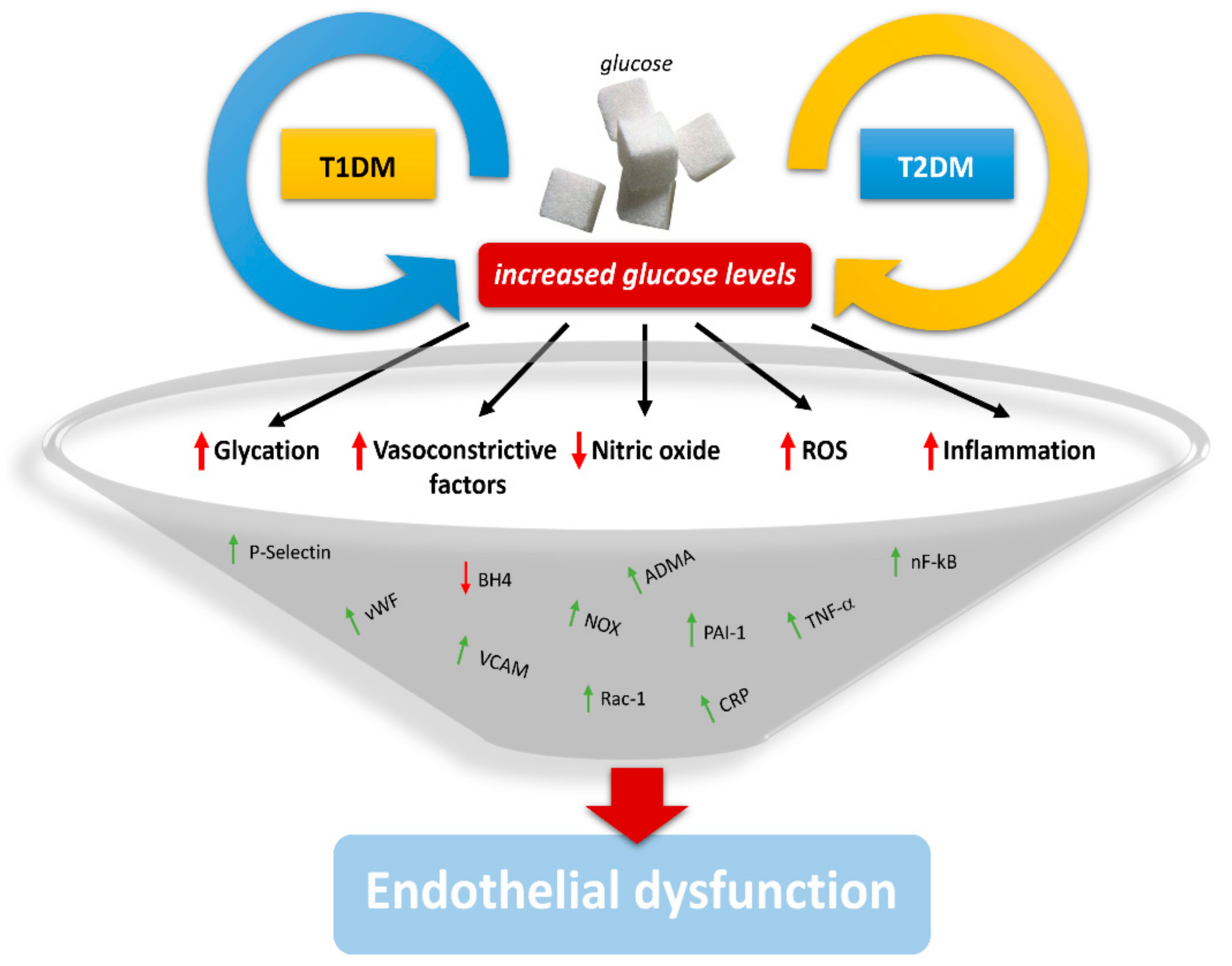{kind=link}
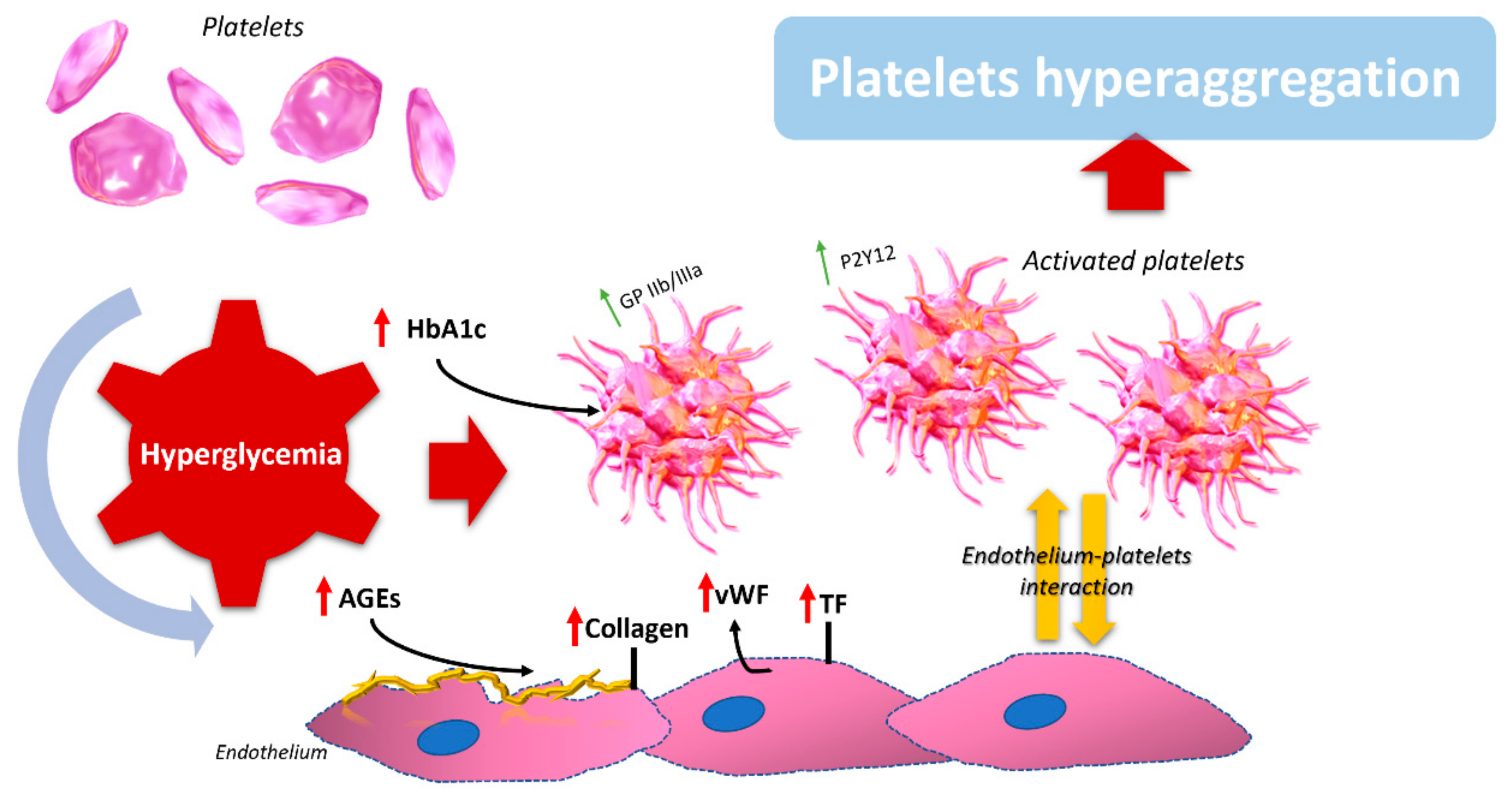{kind=link}
1. Introduction
2. Endothelial Dysfunction and Diabetes
Pathophysiology and Molecular Signaling in Diabetic Endothelial Dysfunction
3. Platelet Aggregation in Diabetes Mellitus
3.1. Biochemical Factors in Diabetic Platelet Dysfunction
3.2. Insulin Effects on Platelets in Diabetes
3.3. Effects of Oxidative Stress and Inflammation on Platelet Function
3.4. Clinical Implications of Platelet Hyperactivity in Diabetes
4. Interactions between Endothelial Dysfunction and Platelet Hyperaggregation in Diabetes
5. Conclusions and Recommendations
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DM | Diabetes Mellitus |
| CVD | Cardiovascular Disease |
| WHO | World Health Organization |
| eNOS | endothelial nitric oxide synthase |
| NO | nitric oxide |
| VCAM-1 | vascular cell adhesion molecule |
| vWF | von Willebrand factor |
| CRP | C-reactive protein |
| TNF | tumor necrosis factor |
| Ang II | angiotensin II |
| PAI-1 | plasminogen activator inhibitor-1 |
| ADMA | asymmetric dimethyl arginine |
| LDL | low-density lipoprotein |
| HDL | high-density lipoprotein |
| VLDL | very low-density lipoprotein |
| NADPH | nicotinamide adenine dinucleotide phosphate oxidase |
| XO | xanthine oxidase |
| GTP | guanosine triphosphate |
| EC | endothelial cell |
| SREBP1 | sterol regulatory element-binding protein 1 |
| CD | cluster of differentiation |
| PKC | protein kinase C |
| HbA1c | glycated hemoglobin |
| AGEs | advanced glycation end products |
| RAGE | AGE receptor |
| GP | glycoprotein |
| P2Y12 | purinergic signaling |
| cAMP | cyclic adenosine monophosphate |
| VASP-P | phosphorylation-dependent vasodilator-stimulated phosphoprotein |
| PK | protein kinase |
| IR | insulin receptor |
| IGF-1 | insulin-like growth factor 1 |
| Mac-1 | macrophage-1 antigen |
| TF | tissue factor |
| COX | cyclooxygenase |
| TXA | thromboxane |
| HTPR | high on-treatment platelet reactivity |
| ADP | adenosine diphosphate |
| ACS | coronary syndrome |
| MI | myocardial infarction |
| PCI | percutaneous coronary intervention |
| DAT | dual antiplatelet therapy |
| PGI2 | prostacyclin |
| Gi | G-protein inhibitory |
References
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Vijan, S. In the clinic. Type 2 diabetes. Ann. Intern. Med. 2010, 152, ITC31–15. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Carrizzo, A.; Ilardi, F.; Damato, A.; Ambrosio, M.; Madonna, M.; Trimarco, V.; Marino, M.; De Angelis, E.; Settembrini, S.; et al. Rac1 modulates endothelial function and platelet aggregation in diabetes mellitus. J. Am. Heart Assoc. 2018, 7, e007322. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.M.; Stefanadis, C. Diabetes mellitus and vascular endothelial dysfunction: Current perspectives. Curr. Vasc. Pharmacol. 2012, 10, 19–32. [Google Scholar] [CrossRef] [PubMed]
- King, H.; Aubert, R.E.; Herman, W.H. Global burden of diabetes, 1995–2025: Prevalence, numerical estimates, and projections. Diabetes Care 1998, 21, 1414–1431. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, C.; Gentile, M.T.; Aretini, A.; Marino, G.; Poulet, R.; Maffei, A.; Passarelli, F.; Landolfi, A.; Vasta, A.; Lembo, G. A novel mechanism of action for statins against diabetes-induced oxidative stress. Diabetologia 2007, 50, 874–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meigs, J.B.; Wilson, P.W.; Fox, C.S.; Vasan, R.S.; Nathan, D.M.; Sullivan, L.M.; D’Agostino, R.B. Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease. J. Clin. Endocrinol. Metab. 2006, 91, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Kampoli, A.M.; Tousoulis, D.; Briasoulis, A.; Latsios, G.; Papageorgiou, N.; Stefanadis, C. Potential pathogenic inflammatory mechanisms of endothelial dysfunction induced by type 2 diabetes mellitus. Curr. Pharm. Des. 2011, 17, 4147–4158. [Google Scholar] [CrossRef] [PubMed]
- Carrizzo, A.; Vecchione, C.; Damato, A.; di Nonno, F.; Ambrosio, M.; Pompeo, F.; Cappello, E.; Capocci, L.; Peruzzi, M.; Valenti, V.; et al. Rac1 pharmacological inhibition rescues human endothelial dysfunction. J. Am. Heart Assoc. 2017, 6, e004746. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, C.; Aretini, A.; Marino, G.; Bettarini, U.; Poulet, R.; Maffei, A.; Sbroggio, M.; Pastore, L.; Gentile, M.T.; Notte, A.; et al. Selective Rac-1 inhibition protects from diabetes-induced vascular injury. Circ. Res. 2006, 98, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Nitric oxide, oxidative excess, and vascular complications of diabetes mellitus. Curr. Hypertens. Rep. 2004, 6, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Sobel, B.E.; Schneider, D.J. Platelet function, coagulopathy, and impaired fibrinolysis in diabetes. Cardiol. Clin. 2004, 22, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Fuller, J.H.; Keen, H.; Jarrett, R.J.; Omer, T.; Meade, T.W.; Chakrabarti, R.; North, W.R.; Stirling, Y. Haemostatic variables associated with diabetes and its complications. Br. Med. J. 1979, 2, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.C.; Grant, P.J. The fibrinolytic system in diabetes mellitus. Diabet Med. 1991, 8, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Puca, A.A.; Carrizzo, A.; Ferrario, A.; Villa, F.; Vecchione, C. Endothelial nitric oxide synthase, vascular integrity and human exceptional longevity. Immun. Ageing 2012, 9, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrizzo, A.; Ambrosio, M.; Damato, A.; Madonna, M.; Storto, M.; Capocci, L.; Campiglia, P.; Sommella, E.; Trimarco, V.; Rozza, F.; et al. Morus alba extract modulates blood pressure homeostasis through enos signaling. Mol. Nutr. Food Res. 2016, 60, 2304–2311. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meigs, J.B.; Hu, F.B.; Rifai, N.; Manson, J.E. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 2004, 291, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Festa, A.; D’Agostino, R., Jr.; Tracy, R.P.; Haffner, S.M.; Insulin Resistance Atherosclerosis Study. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: The insulin resistance atherosclerosis study. Diabetes 2002, 51, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Vischer, U.M.; Emeis, J.J.; Bilo, H.J.; Stehouwer, C.D.; Thomsen, C.; Rasmussen, O.; Hermansen, K.; Wollheim, C.B.; Ingerslev, J. Von willebrand factor (vWf) as a plasma marker of endothelial activation in diabetes: Improved reliability with parallel determination of the vwf propeptide (vWf:AgII). Thromb. Haemost. 1998, 80, 1002–1007. [Google Scholar] [PubMed]
- Balletshofer, B.M.; Rittig, K.; Enderle, M.D.; Volk, A.; Maerker, E.; Jacob, S.; Matthaei, S.; Rett, K.; Haring, H.U. Endothelial dysfunction is detectable in young normotensive first-degree relatives of subjects with type 2 diabetes in association with insulin resistance. Circulation 2000, 101, 1780–1784. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.E.; Arora, S.; Saouaf, R.; Lim, S.C.; Smakowski, P.; Park, J.Y.; King, G.L.; LoGerfo, F.W.; Horton, E.S.; Veves, A. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes 1999, 48, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Klein, R.; Sharrett, A.R.; Schmidt, M.I.; Pankow, J.S.; Couper, D.J.; Klein, B.E.; Hubbard, L.D.; Duncan, B.B.; ARIC Investigators. Retinal arteriolar narrowing and risk of diabetes mellitus in middle-aged persons. JAMA 2002, 287, 2528–2533. [Google Scholar] [CrossRef] [PubMed]
- Picchi, A.; Capobianco, S.; Qiu, T.; Focardi, M.; Zou, X.; Cao, J.M.; Zhang, C. Coronary microvascular dysfunction in diabetes mellitus: A review. World J. Cardiol. 2010, 2, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical update: Cardiovascular disease in diabetes mellitus: Atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus—Mechanisms, management, and clinical considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, K.E.; Celermajer, D.S.; Georgakopoulos, D.; Hatcher, G.; Betteridge, D.J.; Deanfield, J.E. Impairment of endothelium-dependent dilation is an early event in children with familial hypercholesterolemia and is related to the lipoprotein(a) level. J. Clin. Investig. 1994, 93, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Matheus, A.S.; Tannus, L.R.; Cobas, R.A.; Palma, C.C.; Negrato, C.A.; Gomes, M.B. Impact of diabetes on cardiovascular disease: An update. Int. J. Hypertens. 2013, 2013, 653789. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.P. Hyperglycemic endothelial dysfunction: Does it happen and does it matter? J. Thorac. Dis. 2015, 7, 1693–1695. [Google Scholar] [PubMed]
- Pieper, G.M.; Meier, D.A.; Hager, S.R. Endothelial dysfunction in a model of hyperglycemia and hyperinsulinemia. Am. J. Physiol. 1995, 269, H845–H850. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G. Endothelial function and oxidant stress. Clin. Cardiol. 1997, 20, II-11-17. [Google Scholar] [PubMed]
- Forgione, M.A.; Loscalzo, J. Oxidant stress as a critical determinant of endothelial function. Drug News Perspect. 2000, 13, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.W.; Baldwin, S.N. L-arginine in the management of cardiovascular diseases. Ann. Pharmacother. 2001, 35, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Klawitter, J.; Hildreth, K.L.; Christians, U.; Kohrt, W.M.; Moreau, K.L. A relative L-arginine deficiency contributes to endothelial dysfunction across the stages of the menopausal transition. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Heunisch, F.; Chaykovska, L.; von Einem, G.; Alter, M.; Dschietzig, T.; Kretschmer, A.; Kellner, K.H.; Hocher, B. Adma predicts major adverse renal events in patients with mild renal impairment and/or diabetes mellitus undergoing coronary angiography. Medicine 2017, 96, e6065. [Google Scholar] [CrossRef] [PubMed]
- Honing, M.L.; Morrison, P.J.; Banga, J.D.; Stroes, E.S.; Rabelink, T.J. Nitric oxide availability in diabetes mellitus. Diabetes Metab. Rev. 1998, 14, 241–249. [Google Scholar] [CrossRef]
- Harrison, D.G. Cellular and molecular mechanisms of endothelial cell dysfunction. J. Clin. Investig. 1997, 100, 2153–2157. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Conti, V.; Damato, A.; Ambrosio, M.; Puca, A.A.; Sciarretta, S.; Frati, G.; Vecchione, C.; Carrizzo, A. Targeting nitric oxide with natural derived compounds as a therapeutic strategy in vascular diseases. Oxid. Med. Cell. Longev. 2016, 2016, 7364138. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hai, C.X. Ros acts as a double-edged sword in the pathogenesis of type 2 diabetes mellitus: Is Nrf2 a potential target for the treatment? Mini Rev. Med. Chem. 2011, 11, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Amodio, G.; Moltedo, O.; Faraonio, R.; Remondelli, P. Targeting the endoplasmic reticulum unfolded protein response to counteract the oxidative stress-induced endothelial dysfunction. Oxid. Med. Cell. Longev. 2018, 2018, 4946289. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, S.; Landmesser, U.; Dikalov, S.; Bredt, M.; Gamez, G.; Tatge, H.; Reepschlager, N.; Hornig, B.; Drexler, H.; Harrison, D.G. Electron spin resonance characterization of vascular xanthine and nad(p)h oxidase activity in patients with coronary artery disease: Relation to endothelium-dependent vasodilation. Circulation 2003, 107, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Benamer, H.; Hugel, B.; Benessiano, J.; Steg, P.G.; Freyssinet, J.M.; Tedgui, A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 2000, 101, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, K.F.; Pietrani, N.T.; Fernandes, A.P.; Bosco, A.A.; de Sousa, M.C.R.; de Fatima Oliveira Silva, I.; Silveira, J.N.; Campos, F.M.F.; Gomes, K.B. Circulating microparticles levels are increased in patients with diabetic kidney disease: A case-control research. Clin. Chim. Acta 2018, 479, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, E.; Chen, L.; Zhang, Y.; Wei, F.; Liu, J.; Deng, H.; Wang, Y. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating srebp1. Nature 2015, 524, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Amodio, G.; Margarucci, L.; Moltedo, O.; Casapullo, A.; Remondelli, P. Identification of cysteine ubiquitylation sites on the Sec23A protein of the COPII complex required for vesicle formation from the ER. Open Biochem. J. 2017, 11, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002, 106, 2781–2786. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.A.; Liu, Z.J.; Xiao, M.; Chen, H.; Goldstein, L.J.; Buerk, D.G.; Nedeau, A.; Thom, S.R.; Velazquez, O.C. Diabetic impairments in no-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J. Clin. Investig. 2007, 117, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.I.; Erbas, T.; Park, T.S.; Nolan, R.; Pittenger, G.L. Platelet dysfunction in type 2 diabetes. Diabetes Care 2001, 24, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Maiello, M.; Boeri, D.; Podesta, F.; Cagliero, E.; Vichi, M.; Odetti, P.; Adezati, L.; Lorenzi, M. Increased expression of tissue plasminogen activator and its inhibitor and reduced fibrinolytic potential of human endothelial cells cultured in elevated glucose. Diabetes 1992, 41, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Ang, L.; Palakodeti, V.; Khalid, A.; Tsimikas, S.; Idrees, Z.; Tran, P.; Clopton, P.; Zafar, N.; Bromberg-Marin, G.; Keramati, S.; et al. Elevated plasma fibrinogen and diabetes mellitus are associated with lower inhibition of platelet reactivity with clopidogrel. J. Am. Coll. Cardiol. 2008, 52, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Eguchi, H.; Manaka, H.; Igarashi, K.; Kato, T.; Sekikawa, A. Impaired glucose tolerance is a risk factor for cardiovascular disease, but not impaired fasting glucose. The funagata diabetes study. Diabetes Care 1999, 22, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.J.; Scheen, A.J. Glucose metabolism and the postprandial state. Eur. J. Clin. Investig. 1999, 29 (Suppl. 2), 1–6. [Google Scholar] [CrossRef]
- Yngen, M.; Ostenson, C.G.; Li, N.; Hjemdahl, P.; Wallen, N.H. Acute hyperglycemia increases soluble P-selectin in male patients with mild diabetes mellitus. Blood Coagul. Fibrinolysis 2001, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Wiek, I.; Stepien, E.; Zmudka, K.; Tracz, W. Hyperglycemia is associated with enhanced thrombin formation, platelet activation, and fibrin clot resistance to lysis in patients with acute coronary syndrome. Diabetes Care 2008, 31, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Vaidyula, V.R.; Rao, A.K.; Mozzoli, M.; Homko, C.; Cheung, P.; Boden, G. Effects of hyperglycemia and hyperinsulinemia on circulating tissue factor procoagulant activity and platelet cd40 ligand. Diabetes 2006, 55, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Keating, F.K.; Sobel, B.E.; Schneider, D.J. Effects of increased concentrations of glucose on platelet reactivity in healthy subjects and in patients with and without diabetes mellitus. Am. J. Cardiol. 2003, 92, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Assert, R.; Scherk, G.; Bumbure, A.; Pirags, V.; Schatz, H.; Pfeiffer, A.F. Regulation of protein kinase c by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia 2001, 44, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Yngen, M.; Norhammar, A.; Hjemdahl, P.; Wallen, N.H. Effects of improved metabolic control on platelet reactivity in patients with type 2 diabetes mellitus following coronary angioplasty. Diabetes Vasc. Dis. Res. 2006, 3, 52–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhlestein, J.B.; Anderson, J.L.; Horne, B.D.; Lavasani, F.; Allen Maycock, C.A.; Bair, T.L.; Pearson, R.R.; Carlquist, J.F.; Intermountain Heart Collaborative Study (IHCS) Group. Effect of fasting glucose levels on mortality rate in patients with and without diabetes mellitus and coronary artery disease undergoing percutaneous coronary intervention. Am. Heart J. 2003, 146, 351–358. [Google Scholar] [CrossRef]
- Corpus, R.A.; George, P.B.; House, J.A.; Dixon, S.R.; Ajluni, S.C.; Devlin, W.H.; Timmis, G.C.; Balasubramaniam, M.; O’Neill, W.W. Optimal glycemic control is associated with a lower rate of target vessel revascularization in treated type ii diabetic patients undergoing elective percutaneous coronary intervention. J. Am. Coll. Cardiol. 2004, 43, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Schmidt, A.M. Protein glycation: A firm link to endothelial cell dysfunction. Circ. Res. 2004, 95, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Beck, W.; Deppisch, R.; Marshall, S.M.; Hoenich, N.A.; Thompson, M.G. Advanced glycation end products elicit externalization of phosphatidylserine in a subpopulation of platelets via 5-HT2A/2C receptors. Am. J. Physiol. Cell Physiol. 2007, 293, C328–C336. [Google Scholar] [CrossRef] [PubMed]
- Winocour, P.D.; Watala, C.; Perry, D.W.; Kinlough-Rathbone, R.L. Decreased platelet membrane fluidity due to glycation or acetylation of membrane proteins. Thromb. Haemost. 1992, 68, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Watala, C.; Boncer, M.; Golanski, J.; Koziolkiewcz, W.; Trojanowski, Z.; Walkowiak, B. Platelet membrane lipid fluidity and intraplatelet calcium mobilization in type 2 diabetes mellitus. Eur. J. Haematol. 1998, 61, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Tschoepe, D. The activated megakaryocyte-platelet-system in vascular disease: Focus on diabetes. Semin. Thromb. Hemost. 1995, 21, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.A.; Mocking, A.I.; Feijge, M.A.; Gorter, G.; van Haeften, T.W.; Heemskerk, J.W.; Akkerman, J.W. Platelet inhibition by insulin is absent in type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Hechler, B.; Cattaneo, M.; Gachet, C. The p2 receptors in platelet function. Semin. Thromb. Hemost. 2005, 31, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Alexandru, N.; Popov, D.; Sbarcea, A.; Amuzescu, M. Platelet free cytosolic calcium concentration during ageing of type 2 diabetic patients. Platelets 2007, 18, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Pedreno, J.; Hurt-Camejo, E.; Wiklund, O.; Badimon, L.; Masana, L. Platelet function in patients with familial hypertriglyceridemia: Evidence that platelet reactivity is modulated by apolipoprotein e content of very-low-density lipoprotein particles. Metabolism 2000, 49, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Drew, B.G.; Ono, A.; Duffy, S.J.; Gordon, M.V.; Schoenwaelder, S.M.; Sviridov, D.; Cooper, M.E.; Kingwell, B.A.; Jackson, S.P. Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation 2009, 120, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, G.; Rabini, R.A.; Bacchetti, T.; Vignini, A.; Salvolini, E.; Ravaglia, F.; Curatola, G.; Mazzanti, L. Glycated low density lipoproteins modify platelet properties: A compositional and functional study. J. Clin. Endocrinol. Metab. 2002, 87, 2180–2184. [Google Scholar] [CrossRef] [PubMed]
- Falcon, C.; Pfliegler, G.; Deckmyn, H.; Vermylen, J. The platelet insulin receptor: Detection, partial characterization, and search for a function. Biochem. Biophys. Res. Commun. 1988, 157, 1190–1196. [Google Scholar] [CrossRef]
- Hwang, D.L.; Yen, C.F.; Nadler, J.L. Insulin increases intracellular magnesium transport in human platelets. J. Clin. Endocrinol. Metab. 1993, 76, 549–553. [Google Scholar] [PubMed]
- Trovati, M.; Anfossi, G.; Massucco, P.; Mattiello, L.; Costamagna, C.; Piretto, V.; Mularoni, E.; Cavalot, F.; Bosia, A.; Ghigo, D. Insulin stimulates nitric oxide synthesis in human platelets and, through nitric oxide, increases platelet concentrations of both guanosine-3′, 5′-cyclic monophosphate and adenosine-3′, 5′-cyclic monophosphate. Diabetes 1997, 46, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.A.; Eybrechts, K.L.; Mocking, A.I.; Kroner, C.; Akkerman, J.W. IRS-1 mediates inhibition of Ca2+ mobilization by insulin via the inhibitory G-protein Gi. J. Biol. Chem. 2004, 279, 3254–3264. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Hers, I. Insulin/IGF-1 hybrid receptor expression on human platelets: Consequences for the effect of insulin on platelet function. J. Thromb. Haemost. 2009, 7, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Hers, I. Insulin-like growth factor-1 potentiates platelet activation via the Irs/PI3Kalpha pathway. Blood 2007, 110, 4243–4252. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, K.; Nozaki, H.; Arimori, S. Reduction of platelet aggregation induced by euglycaemic insulin clamp. Diabetologia 1987, 30, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Trovati, M.; Anfossi, G.; Cavalot, F.; Massucco, P.; Mularoni, E.; Emanuelli, G. Insulin directly reduces platelet sensitivity to aggregating agents. Studies in vitro and in vivo. Diabetes 1988, 37, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Westerbacka, J.; Yki-Jarvinen, H.; Turpeinen, A.; Rissanen, A.; Vehkavaara, S.; Syrjala, M.; Lassila, R. Inhibition of platelet-collagen interaction: An in vivo action of insulin abolished by insulin resistance in obesity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Horigome, H.; Tanaka, K.; Nakata, Y.; Ohkawara, K.; Katayama, Y.; Matsui, A. Impact of weight reduction on production of platelet-derived microparticles and fibrinolytic parameters in obesity. Thromb. Res. 2007, 119, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davi, G.; Guagnano, M.T.; Ciabattoni, G.; Basili, S.; Falco, A.; Marinopiccoli, M.; Nutini, M.; Sensi, S.; Patrono, C. Platelet activation in obese women: Role of inflammation and oxidant stress. JAMA 2002, 288, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Basili, S.; Pacini, G.; Guagnano, M.T.; Manigrasso, M.R.; Santilli, F.; Pettinella, C.; Ciabattoni, G.; Patrono, C.; Davi, G. Insulin resistance as a determinant of platelet activation in obese women. J. Am. Coll. Cardiol. 2006, 48, 2531–2538. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Ishida, T.; Ono, N.; Matsuura, H.; Watanabe, M.; Kajiyama, G.; Kambe, M.; Oshima, T. Effects of insulin on calcium metabolism and platelet aggregation. Hypertension 1996, 28, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Bernardo, E.; Ramirez, C.; Costa, M.A.; Sabate, M.; Jimenez-Quevedo, P.; Hernandez, R.; Moreno, R.; Escaned, J.; Alfonso, F.; et al. Insulin therapy is associated with platelet dysfunction in patients with type 2 diabetes mellitus on dual oral antiplatelet treatment. J. Am. Coll. Cardiol. 2006, 48, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J.; El Tahir, K.E.; Reckless, J.P.; Williams, K.I. Platelets from diabetic subjects show diminished sensitivity to prostacyclin. Eur. J. Clin. Investig. 1982, 12, 395–398. [Google Scholar] [CrossRef]
- Anfossi, G.; Mularoni, E.M.; Burzacca, S.; Ponziani, M.C.; Massucco, P.; Mattiello, L.; Cavalot, F.; Trovati, M. Platelet resistance to nitrates in obesity and obese niddm, and normal platelet sensitivity to both insulin and nitrates in lean niddm. Diabetes Care 1998, 21, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Vaidyula, V.R.; Boden, G.; Rao, A.K. Platelet and monocyte activation by hyperglycemia and hyperinsulinemia in healthy subjects. Platelets 2006, 17, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Redondo, P.C.; Jardin, I.; Hernandez-Cruz, J.M.; Pariente, J.A.; Salido, G.M.; Rosado, J.A. Hydrogen peroxide and peroxynitrite enhance Ca2+ mobilization and aggregation in platelets from type 2 diabetic patients. Biochem. Biophys. Res. Commun. 2005, 333, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E. Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, s11–s16. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Redondo, P.C.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Endogenously generated reactive oxygen species reduce pmca activity in platelets from patients with non-insulin-dependent diabetes mellitus. Platelets 2006, 17, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Davi, G.; Ciabattoni, G.; Consoli, A.; Mezzetti, A.; Falco, A.; Santarone, S.; Pennese, E.; Vitacolonna, E.; Bucciarelli, T.; Costantini, F.; et al. In vivo formation of 8-Iso-Prostaglandin F2alpha and platelet activation in diabetes mellitus: Effects of improved metabolic control and vitamin E supplementation. Circulation 1999, 99, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.J.; Gopaul, N.; Davies, I.R.; Hughes, D.A.; Carrier, M.J. Plasma F2 isoprostanes: Direct evidence of increased free radical damage during acute hyperglycemia in type 2 diabetes. Diabetes Care 2002, 25, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, G.; Wascher, T.C.; Kostner, G.M.; Graier, W.F. Alterations in platelet Ca2+ signalling in diabetic patients is due to increased formation of superoxide anions and reduced nitric oxide production. Diabetologia 1999, 42, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Wautier, J.L.; Stern, D. Activation of receptor for advanced glycation end products: A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 1999, 84, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The rage axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ. Res. 2010, 106, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Gawlowski, T.; Stratmann, B.; Ruetter, R.; Buenting, C.E.; Menart, B.; Weiss, J.; Vlassara, H.; Koschinsky, T.; Tschoepe, D. Advanced glycation end products strongly activate platelets. Eur. J. Nutr. 2009, 48, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Li, W.; Silverstein, R.L. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012, 119, 6136–6144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawlowski, T.; Stratmann, B.; Stirban, A.O.; Negrean, M.; Tschoepe, D. AGEs and methylglyoxal induce apoptosis and expression of Mac-1 on neutrophils resulting in platelet-neutrophil aggregation. Thromb. Res. 2007, 121, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Suehiro, A.; Higasa, S.; Namba, M.; Kakishita, E. Enhancing effect of advanced glycation end products on serotonin-induced platelet aggregation in patients with diabetes mellitus. Thromb. Res. 2002, 107, 319–323. [Google Scholar] [CrossRef]
- Negrean, M.; Stirban, A.; Stratmann, B.; Gawlowski, T.; Horstmann, T.; Gotting, C.; Kleesiek, K.; Mueller-Roesel, M.; Koschinsky, T.; Uribarri, J.; et al. Effects of low- and high-advanced glycation endproduct meals on macro- and microvascular endothelial function and oxidative stress in patients with type 2 diabetes mellitus. Am. J. Clin. Nutr. 2007, 85, 1236–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinetti, G.; Kraenkel, N.; Emanueli, C.; Madeddu, P. Diabetes and vessel wall remodelling: From mechanistic insights to regenerative therapies. Cardiovasc. Res. 2008, 78, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.J. Factors contributing to increased platelet reactivity in people with diabetes. Diabetes Care 2009, 32, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Linden, E.; Cai, W.; He, J.C.; Xue, C.; Li, Z.; Winston, J.; Vlassara, H.; Uribarri, J. Endothelial dysfunction in patients with chronic kidney disease results from advanced glycation end products (AGE)-mediated inhibition of endothelial nitric oxide synthase through rage activation. Clin. J. Am. Soc. Nephrol. 2008, 3, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Guthikonda, S.; Lev, E.I.; Patel, R.; DeLao, T.; Bergeron, A.L.; Dong, J.F.; Kleiman, N.S. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin. J. Thromb. Haemost. 2007, 5, 490–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthikonda, S.; Alviar, C.L.; Vaduganathan, M.; Arikan, M.; Tellez, A.; DeLao, T.; Granada, J.F.; Dong, J.F.; Kleiman, N.S.; Lev, E.I. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002, 324, 71–86. [Google Scholar] [CrossRef] [Green Version]
- Beigel, R.; Hod, H.; Fefer, P.; Asher, E.; Novikov, I.; Shenkman, B.; Savion, N.; Varon, D.; Matetzky, S. Relation of aspirin failure to clinical outcome and to platelet response to aspirin in patients with acute myocardial infarction. Am. J. Cardiol. 2011, 107, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Krasopoulos, G.; Brister, S.J.; Beattie, W.S.; Buchanan, M.R. Aspirin “resistance” and risk of cardiovascular morbidity: Systematic review and meta-analysis. BMJ 2008, 336, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Eikelboom, J.W.; Hirsh, J.; Weitz, J.I.; Johnston, M.; Yi, Q.; Yusuf, S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation 2002, 105, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- Breet, N.J.; van Werkum, J.W.; Bouman, H.J.; Kelder, J.C.; Ten Berg, J.M.; Hackeng, C.M. High on-aspirin platelet reactivity as measured with aggregation-based, cyclooxygenase-1 inhibition sensitive platelet function tests is associated with the occurrence of atherothrombotic events. J. Thromb. Haemost. 2010, 8, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Reny, J.L.; De Moerloose, P.; Dauzat, M.; Fontana, P. Use of the PFA-100 closure time to predict cardiovascular events in aspirin-treated cardiovascular patients: A systematic review and meta-analysis. J. Thromb. Haemost. 2008, 6, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Eskandarian, R.; Razavi, M.; Fattah, A.; Ghods, K.; Forozeshfard, M. Prevalence of aspirin resistance in patients with type ii diabetes: A descriptive-analytical study. Int. J. Clin. Pharmacol. Ther. 2017, 55, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.S.; Silver, R.J.; Aaronson, A.; Abrahamson, M.; Goldfine, A.B. Comparison of aspirin resistance in type 1 versus type 2 diabetes mellitus. Am. J. Cardiol. 2006, 97, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.M.; Segal, J.; Vaidya, D.; Yanek, L.R.; Herrera-Galeano, J.E.; Bray, P.F.; Moy, T.F.; Becker, L.C.; Faraday, N. Sex differences in platelet reactivity and response to low-dose aspirin therapy. JAMA 2006, 295, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, T.J.; McLean, R.C.; Schulman, S.P.; Kickler, T.S.; Shapiro, E.P.; Conte, J.V.; McNicholas, K.W.; Segal, J.B.; Rade, J.J. Effects of aspirin responsiveness and platelet reactivity on early vein graft thrombosis after coronary artery bypass graft surgery. J. Am. Coll. Cardiol. 2011, 57, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Jurasz, P.; Hollenberg, M.D.; Radomski, M.W. Mechanisms of action of proteinase-activated receptor agonists on human platelets. Br. J. Pharmacol. 2002, 135, 1123–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, S.; Zhao, F.; Mehta, S.R.; Chrolavicius, S.; Tognoni, G.; Fox, K.K. Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial, I. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-Segment elevation. N. Engl. J. Med. 2001, 345, 494–502. [Google Scholar] [PubMed]
- Bhatt, D.L.; Fox, K.A.; Hacke, W.; Berger, P.B.; Black, H.R.; Boden, W.E.; Cacoub, P.; Cohen, E.A.; Creager, M.A.; Easton, J.D.; et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N. Engl. J. Med. 2006, 354, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bhatt, D.L.; Dunn, E.S.; Shi, C.; Caro, J.J.; Mahoney, E.M.; Gabriel, S.; Jackson, J.D.; Topol, E.J.; Cohen, D.J. Cost-effectiveness of clopidogrel plus aspirin versus aspirin alone for secondary prevention of cardiovascular events: Results from the charisma trial. Value Health 2009, 12, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Steinhubl, S.R.; Bhatt, D.L.; Berger, P.B.; Shao, M.; Mak, K.H.; Fox, K.A.; Montalescot, G.; Weber, M.A.; Haffner, S.M.; et al. Clinical outcomes of patients with diabetic nephropathy randomized to clopidogrel plus aspirin versus aspirin alone (a post hoc analysis of the clopidogrel for high atherothrombotic risk and ischemic stabilization, management, and avoidance [charisma] trial). Am. J. Cardiol. 2009, 103, 1359–1363. [Google Scholar] [PubMed]
- Pignone, M.; Alberts, M.J.; Colwell, J.A.; Cushman, M.; Inzucchi, S.E.; Mukherjee, D.; Rosenson, R.S.; Williams, C.D.; Wilson, P.W.; Kirkman, M.S.; et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: A position statement of the american diabetes association, a scientific statement of the american heart association, and an expert consensus document of the american college of cardiology foundation. Diabetes Care 2010, 33, 1395–1402. [Google Scholar] [PubMed]
- Gryglewski, R.J.; Botting, R.M.; Vane, J.R. Mediators produced by the endothelial cell. Hypertension 1988, 12, 530–548. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Anggard, E.E.; Botting, R.M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990, 323, 27–36. [Google Scholar] [PubMed]
- Klaff, L.J.; Kernoff, L.; Vinik, A.I.; Jackson, W.P.; Jacobs, P. Sulfonylureas and platelet function. Am. J. Med. 1981, 70, 627–630. [Google Scholar] [CrossRef]
- Jakobs, K.H.; Aktories, K.; Minuth, M.; Schultz, G. Inhibition of adenylate cyclase. Adv. Cyclic Nucleotide Protein Phosphorylation Res. 1985, 19, 137–150. [Google Scholar] [PubMed]
- Lazarowski, E.R.; Winegar, D.A.; Nolan, R.D.; Oberdisse, E.; Lapetina, E.G. Effect of protein kinase A on inositide metabolism and rap 1 G-protein in human erythroleukemia cells. J. Biol. Chem. 1990, 265, 13118–13123. [Google Scholar] [PubMed]
- Gerrard, J.M.; Stuart, M.J.; Rao, G.H.; Steffes, M.W.; Mauer, S.M.; Brown, D.M.; White, J.G. Alteration in the balance of prostaglandin and thromboxane synthesis in diabetic rats. J. Lab. Clin. Med. 1980, 95, 950–958. [Google Scholar] [PubMed]
- Harrison, H.E.; Reece, A.H.; Johnson, M. Effect of insulin treatment on prostacyclin in experimental diabetes. Diabetologia 1980, 18, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Ylikorkala, O.; Kaila, J.; Viinikka, L. Prostacyclin and thromboxane in diabetes. Br. Med. J. (Clin. Res. Ed.) 1981, 283, 1148–1150. [Google Scholar] [CrossRef]
- Bucala, R.; Tracey, K.J.; Cerami, A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J. Clin. Investig. 1991, 87, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.H.; Azhar, S.; Hoffman, B.B. Inactivation of endothelial derived relaxing factor by oxidized lipoproteins. J. Clin. Investig. 1992, 89, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Akai, T.; Naka, K.; Okuda, K.; Takemura, T.; Fujii, S. Decreased sensitivity of platelets to prostacyclin in patients with diabetes mellitus. Horm. Metab. Res. 1983, 15, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Mazumder, S.; Sinha, A.K. The role of inhibition of nitric oxide synthesis in the aggregation of platelets due to the stimulated production of thromboxane A2. Blood Coagul. Fibrinolysis 2014, 25, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Randriamboavonjy, V.; Fleming, I. Insulin, insulin resistance, and platelet signaling in diabetes. Diabetes Care 2009, 32, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Kageyama, S.; Sakurai, T.; Murakawa, Y.; Aihara, K.; Yokota, K.; Taniguchi, I.; Hashimoto, Y.; Fujita, T.; Tajima, N. Inhibitory effects of insulin on intracellular calcium and aggregatory response of platelets are impaired in hypertensive subjects with insulin resistance. Hypertens. Res. 1997, 20, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Winocour, P.D.; Lopes-Virella, M.; Laimins, M.; Colwell, J.A. Effect of insulin treatment in streptozocin-induced diabetic rats on in vitro platelet function and plasma von willebrand factor activity and factor VIII-related antigen. J. Lab. Clin. Med. 1985, 106, 319–325. [Google Scholar] [PubMed]
- Van Zile, J.; Kilpatrick, M.; Laimins, M.; Sagel, J.; Colwell, J.; Virella, G. Platelet aggregation and release of atp after incubation with soluble immune complexes purified from the serum of diabetic patients. Diabetes 1981, 30, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.; Brett, J.; Radoff, S.; Ogawa, S.; Stern, D.; Vlassara, H. Advanced protein glycosylation induces transendothelial human monocyte chemotaxis and secretion of platelet-derived growth factor: Role in vascular disease of diabetes and aging. Proc. Natl. Acad. Sci. USA 1990, 87, 9010–9014. [Google Scholar] [CrossRef] [PubMed]
- Stirban, A.; Negrean, M.; Stratmann, B.; Gawlowski, T.; Horstmann, T.; Gotting, C.; Kleesiek, K.; Mueller-Roesel, M.; Koschinsky, T.; Uribarri, J.; et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 2006, 29, 2064–2071. [Google Scholar] [CrossRef] [PubMed]
- Hangaishi, M.; Taguchi, J.; Miyata, T.; Ikari, Y.; Togo, M.; Hashimoto, Y.; Watanabe, T.; Kimura, S.; Kurokawa, K.; Ohno, M. Increased aggregation of human platelets produced by advanced glycation end products in vitro. Biochem. Biophys. Res. Commun. 1998, 248, 285–292. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrizzo, A.; Izzo, C.; Oliveti, M.; Alfano, A.; Virtuoso, N.; Capunzo, M.; Di Pietro, P.; Calabrese, M.; De Simone, E.; Sciarretta, S.; et al. The Main Determinants of Diabetes Mellitus Vascular Complications: Endothelial Dysfunction and Platelet Hyperaggregation. Int. J. Mol. Sci. 2018, 19, 2968. https://doi.org/10.3390/ijms19102968
Carrizzo A, Izzo C, Oliveti M, Alfano A, Virtuoso N, Capunzo M, Di Pietro P, Calabrese M, De Simone E, Sciarretta S, et al. The Main Determinants of Diabetes Mellitus Vascular Complications: Endothelial Dysfunction and Platelet Hyperaggregation. International Journal of Molecular Sciences. 2018; 19(10):2968. https://doi.org/10.3390/ijms19102968
Chicago/Turabian StyleCarrizzo, Albino, Carmine Izzo, Marco Oliveti, Antonia Alfano, Nicola Virtuoso, Mario Capunzo, Paola Di Pietro, Mariaconsiglia Calabrese, Eros De Simone, Sebastiano Sciarretta, and et al. 2018. "The Main Determinants of Diabetes Mellitus Vascular Complications: Endothelial Dysfunction and Platelet Hyperaggregation" International Journal of Molecular Sciences 19, no. 10: 2968. https://doi.org/10.3390/ijms19102968
APA StyleCarrizzo, A., Izzo, C., Oliveti, M., Alfano, A., Virtuoso, N., Capunzo, M., Di Pietro, P., Calabrese, M., De Simone, E., Sciarretta, S., Frati, G., Migliarino, S., Damato, A., Ambrosio, M., De Caro, F., & Vecchione, C. (2018). The Main Determinants of Diabetes Mellitus Vascular Complications: Endothelial Dysfunction and Platelet Hyperaggregation. International Journal of Molecular Sciences, 19(10), 2968. https://doi.org/10.3390/ijms19102968