QSAR and Classification Study on Prediction of Acute Oral Toxicity of N-Nitroso Compounds



Abstract
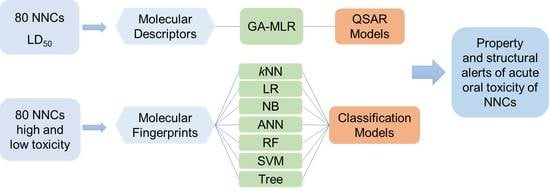
:
1. Introduction
2. Results and Discussion
2.1. QSAR Models
2.1.1. Model Validation
Ntest = 14, Q2ext = 0.7041, R2ext = 0.7195, RMSEtest = 0.2847, Q2F1 = 0.7041, Q2F2 = 0.7032, Q2F3 = 0.7794, CCCtest = 0.8062, (R2ext − R02)/R2ext = 0.0215, |R02 − R’02| = 0.2642.
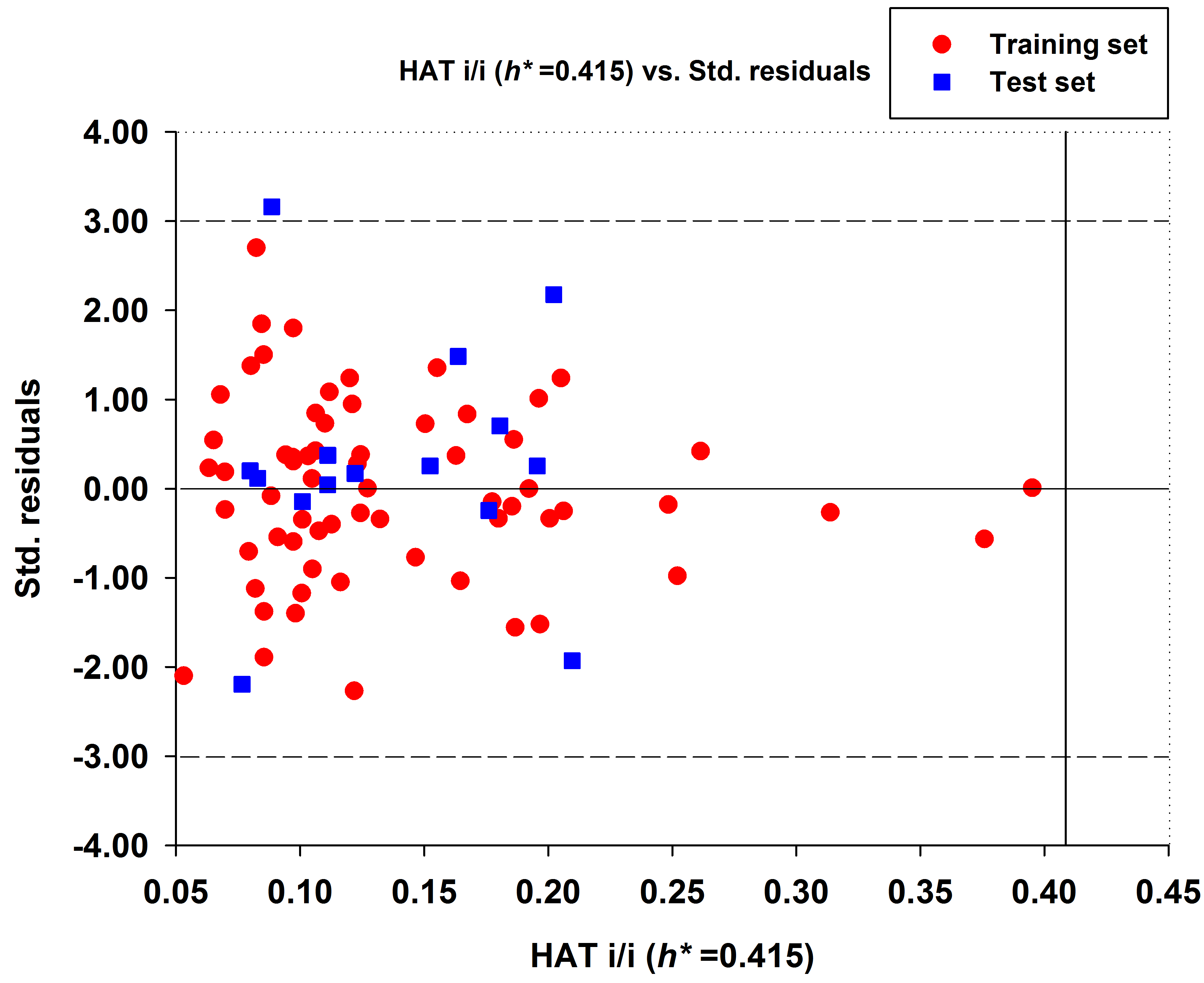
2.1.2. Outlier Analysis of MLR Model
2.1.3. Interpretation of Descriptors in MLR Model
2.2. Classification Models
2.2.1. Data Set Analysis
2.2.2. Performances of 10-Fold Cross-Validation
2.2.3. Performance of External Test Set
2.2.4. Identification of Privileged Substructures as Structural Alerts
3. Materials and Methods
3.1. QSAR Study
3.1.1. Data Preparation
3.1.2. Calculation of Descriptors
3.1.3. QSAR Modeling and Model Evaluation
3.1.4. Application Domain
3.2. Classification Study
3.2.1. Data Preparation
3.2.2. Molecular Fingerprints
3.2.3. Machine Learning Methods
3.2.4. Performance Evaluation
3.2.5. Analysis of Privileged Substructures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lijinsky, W. N-nitroso compounds in the diet. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 1999, 443, 129–138. [Google Scholar] [CrossRef]
- Druckrey, H.; Preussmann, R.; Ivankovic, S.; Schmahl, D. Organotropic carcinogenic effects of 65 various N-nitroso- compounds on BD rats. Z. Krebsforsch. 1967, 69, 103–201. [Google Scholar] [CrossRef] [PubMed]
- Lijinsky, W.; Andrews, A.W. N-nitrosamine mutagenicity using the Salmonella/Mammalian-microsome mutagenicity assay. In Genotoxicology of N-Nitroso Compounds; Rao, T.K., Lijinsky, W., Epler, J.L., Eds.; Plenum Press: New York, NY, USA, 1984; pp. 13–43. [Google Scholar]
- Lijinsky, W. Structure-activity relations in carcinogenesis by N-nitroso compounds. Cancer Metast. Rev. 1987, 6, 301–356. [Google Scholar] [CrossRef]
- Bartsch, H.; Oshima, H.; Pignatelli, B.; Malaveille, C.; Friesen, M. Nitrite-reactive phenols present in smoked foods and amino-sugars formed by the Maillard reaction as precursors of genotoxic arenediazonium ions or nitroso compounds. In Mutagens in Food: Detection and Prevention; Hikoya, H., Ed.; CRC Press: Boca Raton, FL, USA, 1991; pp. 87–100. [Google Scholar]
- Lijinsky, W. Chemistry and Biology of N-Nitroso Compounds; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Yuan, J.T.; Pu, Y.P.; Yin, L.H. Predicting carcinogenicity and understanding the carcinogenic mechanism of N-nitroso compounds using a TOPS-MODE approach. Chem. Res. Toxicol. 2011, 24, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Laires, A.; Gaspar, J.; Borba, H.; Proenca, M.; Monteiro, M.; Rueff, J. Genotoxicity of nitrosated red wine and of the nitrosatable phenolic-compounds present in wine-tyramine, quercetin and malvidine-3-glucoside. Food Chem. Toxicol. 1993, 31, 989–994. [Google Scholar] [CrossRef]
- Gaspar, J.; Laires, A.; Va, S.; Pereira, S.; Mariano, A.; Quina, M.; Rueff, J. Mutagenic activity of glycine upon nitrosation in the presence of chloride and human gastric juice: A possible role in gastric carcinogenesis. Teratog. Carcinog. Mutagen. 1996, 16, 275–286. [Google Scholar] [CrossRef]
- Duarte, M.P.; Laires, A.; Gaspar, J.; Oliveira, J.S.; Rueff, J. Genotoxicity of instant coffee and of some phenolic compounds present in coffee upon nitrosation. Teratog. Carcinog. Mutagen. 2000, 20, 241–249. [Google Scholar] [CrossRef]
- Bartsch, H.; Ohshima, H.; Pignatelli, B. Inhibitors of endogenous nitrosation—Mechanisms and implications in human cancer prevention. Mutat. Res. 1988, 202, 307–324. [Google Scholar] [CrossRef]
- Tratnyek, P.G.; Bylaska, E.J.; Weber, E.J. In silico environmental chemical science: Properties and processes from statistical and computational modelling. Environ. Sci. Process. Impacts 2017, 19, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Card, M.L.; Gomez-Alvarez, V.; Lee, W.; Lynch, D.G.; Orentas, N.S.; Lee, M.T.; Wong, E.M.; Boethling, R.S. History of EPI Suite((TM)) and future perspectives on chemical property estimation in US Toxic Substances Control Act new chemical risk assessments. Environ. Sci. Process. Impacts 2017, 19, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M.; Walker, J.D.; Jaworska, J.S.; Comber, M.; Watts, C.D.; Worth, A.P. Use of QSARs in international decision-making frameworks to predict ecologic effects and environmental fate of chemical substances. Environ. Health Perspect. 2003, 111, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Combes, R.; Grindon, C.; Cronin, M.; Roberts, D.W.; Garrod, J.F. Integrated decision-tree testing strategies for acute systemic toxicity and toxicokinetics with respect to the requirements of the EU REACH legislation. Atla-Altern. Lab. Anim. 2008, 36, 45–63. [Google Scholar]
- Cronin, M.; Madden, J.; Enoch, S.; Roberts, D. Evaluation of categories and read-across for toxicity prediction allowing for regulatory acceptance. In Chemical Toxicity Prediction: Category Formation and ReadAcross; The Royal Society of Chemistry: Cambridge, UK, 2013; pp. 155–167. [Google Scholar]
- Cronin, M.T. (Q)SARs to predict environmental toxicities: Current status and future needs. Environ. Sci. Process. Impacts 2017, 19, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.H.; Zhong, R.G.; Gao, X.M. Pattern recognition data for structure-carcinogenic activity relationship of N-nitroso compounds based upon di-region theory. Environ. Chem. 1987, 6, 1–12. [Google Scholar]
- Luan, F.; Zhang, R.S.; Zhao, C.Y.; Yao, X.J.; Liu, M.C.; Hu, Z.D.; Fan, B.T. Classification of the carcinogenicity of N-nitroso compounds based on support vector machines and linear discriminant analysis. Chem. Res. Toxicol. 2005, 18, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Helguera, A.M.; Gonzalez, M.P.; Dias Soeiro Cordeiro, M.N.; Cabrera Perez, M.A. Quantitative structure—Carcinogenicity relationship for detecting structural alerts in nitroso compounds: Species, rat; Sex, female; Route of administration, Gavage. Chem. Res. Toxicol. 2008, 21, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Helguera, A.M.; Cordeiro, M.N.D.S.; Pérez, M.Á.C.; Combes, R.D.; González, M.P. Quantitative structure carcinogenicity relationship for detecting structural alerts in nitroso-compounds☆Species: Rat; Sex: Male; Route of administration: Water. Toxicol. Appl. Pharm. 2008, 231, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hopfinger, A.J. Application of genetic function approximation to quantitative structure-activity-relationships and quantitative structure-property relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Q.; Hu, J. QSAR study of the acute toxicity to fathead minnow based on a large dataset. SAR QSAR Environ. Res. 2016, 27, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Beware of q(2)! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim, Germany, 2008; Volume 11. [Google Scholar]
- Cassotti, M.; Ballabio, D.; Consonni, V.; Mauri, A.; Tetko, I.V.; Todeschini, R. Prediction of acute aquatic toxicity toward daphnia magna by using the GA-kNN method. Atla-Altern. Lab. Anim. 2014, 42, 31–41. [Google Scholar]
- Zhang, J.; Ji, L.; Liu, W. In Silico Prediction of Cytochrome P450-mediated biotransformations of xenobiotics: A case study of epoxidation. Chem. Res. Toxicol. 2015, 28, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Galvez, J.; Garciadomenech, R.; Dejulianortiz, V.; Soler, R. Topological approach to analgesia. J. Chem. Inf. Comput. Sci. 1994, 34, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Galvez, J.; Garciadomenech, R.; Dejulianortiz, J.V.; Soler, R. Topological approach to drug design. J. Chem. Inf. Comput. Sci. 1995, 35, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Roberto, T.; Consonni, V. Molecular Descriptors for Chemoinformatics. In Methods and Principles in Medicinal Chemistry; Mannhold, R., KubiAnyi, H., Folkers, G., Eds.; WILEY-VCH: Weinheim, Germany, 2009; pp. 617–618. [Google Scholar]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for 3-dimensional structure-directed quantitative structure-activity-relationships I. Partition-coefficients as a measure of hydrophobicity. J. Comput. Chem. 1986, 7, 565–577. [Google Scholar] [CrossRef]
- Xu, C.; Cheng, F.; Chen, L.; Du, Z.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. In silico prediction of chemical Ames mutagenicity. J. Chem. Inf. Model. 2012, 52, 2840–2847. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Cai, Y.; Yang, H.; Zhang, H.; Xue, Y.; Liu, G.; Tang, Y.; Li, W. In silico prediction of chemicals binding to aromatase with machine learning methods. Chem. Res. Toxicol. 2017, 30, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.V. Advantages and disadvantages of using artificial neural networks versus logistic regression for predicting medical outcomes. J. Clin. Epidemiol. 1996, 49, 1225–1231. [Google Scholar] [CrossRef]
- Li, X.; Chen, L.; Cheng, F.; Wu, Z.; Bian, H.; Xu, C.; Li, W.; Liu, G.; Shen, X.; Tang, Y. In silico prediction of chemical acute oral toxicity using multi-classification methods. J. Chem. Inf. Model. 2014, 54, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cheng, F.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. In silico prediction of drug induced liver toxicity using substructure pattern recognition method. Mol. Inform. 2016, 35, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Satpute, R.M.; Hariharakrishnan, J.; Tripathi, H.; Saxena, P.B. Acute toxicity of some synthetic cyanogens in rats and their response to oral treatment with alpha-ketoglutarate. Food Chem. Toxicol. 2009, 47, 2314–2320. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Li, Y.; Song, Y.; Li, D.; Sun, H.; Hou, T. ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. J. Cheminform. 2016, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Zhao, L.J.; Fan, T.J.; Li, S.S.; Zhong, R.G. Investigations on the effect of O-6-benzylguanine on the formation of dG-dC interstrand cross-links induced by chloroethylnitrosoureas in human glioma cells using stable isotope dilution high-performance liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2014, 27, 1253–1262. [Google Scholar] [PubMed]
- Sun, G.H.; Zhang, N.; Zhao, L.J.; Fan, T.J.; Zhang, S.F.; Zhong, R.G. Synthesis and antitumor activity evaluation of a novel combi-nitrosourea prodrug: Designed to release a DNA cross-linking agent and an inhibitor of O6-alkylguanine-DNA alkyltransferase. Bioorg. Med. Chem. 2016, 24, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Fan, T.; Zhao, L.; Zhou, Y.; Zhong, R. The potential of combi-molecules with DNA-damaging function as anticancer agents. Future Med. Chem. 2017, 9, 403–435. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Zhao, L.J.; Zhong, R.G.; Peng, Y.Z. The specific role of O6-methylguanine-DNA methyltransferase inhibitors in cancer chemotherapy. Future Med. Chem. 2018, 16, 1971–1996. [Google Scholar] [CrossRef] [PubMed]
- Ma, I.; Peterlin, L. Role of cyclic tertiary amine bioactivation to reactive iminium species: Structure toxicity relationship. Curr. Drug Metab. 2011, 12, 35–50. [Google Scholar]
- Borgelt, C.; Meinl, T.; Berthold, M. MoSS: A program for molecular substructure mining. In Proceedings of the 1st International Workshop on Open Source Data Mining: Frequent Pattern Mining Implementations, Chicago, IL, USA, 21 August 2005. [Google Scholar]
- TOXNET-ChemIDplus. Available online: https://chem.nlm.nih.gov/chemidplus/ (accessed on 3 June 2017).
- Schweinsberg, F.; Schottkollat, P.; Burkle, G. Change of toxicity and carcinogenicity of N-methyl-N-nitrosobenzylamine in rats by methylsubstitution in phenylresidue. Z. Krebsforsch. 1977, 88, 231–236. [Google Scholar] [CrossRef]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996, 96, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Karabulut, S.; Sizochenko, N.; Orhan, A.; Leszczynski, J. A DFT-based QSAR study on inhibition of human dihydrofolate reductase. J. Mol. Graph. Model. 2016, 70, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Luo, F.; Zeng, Z.; Wen, L.; Xiao, Z.; Bu, H.; Lv, F.; Xu, Z.; Lin, Q. DFT-based quantitative structure-activity relationship studies for antioxidant peptides. Struct. Chem. 2015, 26, 739–747. [Google Scholar] [CrossRef]
- Nendza, M.; Mueller, M.; Wenzel, A. Classification of baseline toxicants for QSAR predictions to replace fish acute toxicity studies. Environ. Sci. Process. Impacts 2017, 19, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Enoch, S.J.; Cronin, M.T.D.; Schultz, T.W.; Madden, J.C. Quantitative and mechanistic read across for predicting the skin sensitization potential of alkenes acting via Michael addition. Chem. Res. Toxicol. 2008, 21, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Pasha, F.A.; Muddassar, M.; Beg, Y.; Cho, S.J. DFT-based de novo QSAR of phenoloxidase inhibitors. Chem. Biol. Drug Des. 2008, 71, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kode Srl. Dragon (Software for Molecular Descriptor Calculation) V 7.0.6. Available online: https://chm.kode-solutions.net/ (accessed on 3 September 2017).
- Onlu, S.; Turker, S.M. Impact of geometry optimization methods on QSAR modelling: A case study for predicting human serum albumin binding affinity. SAR QSAR Environ. Res. 2017, 28, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V.; Maiocchi, A. The K correlation index: Theory development and its application in chemometrics. Chemometr. Intell. Lab. Syst. 1999, 46, 13–29. [Google Scholar] [CrossRef]
- Shi, L.M.; Fang, H.; Tong, W.D.; Wu, J.; Perkins, R.; Blair, R.M.; Branham, W.S.; Dial, S.L.; Moland, C.I.; Sheehan, D.M. QSAR models using a large diverse set of estrogens. J. Chem. Inf. Comput. Sci. 2001, 41, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Schueuermann, G.; Ebert, R.; Chen, J.; Wang, B.; Kuehne, R. External validation and prediction employing the predictive squared correlation coefficient-test set activity mean vs training set activity mean. J. Chem. Inf. Model. 2008, 48, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Ballabio, D.; Todeschini, R. Comments on the definition of the q(2) parameter for QSAR validation. J. Chem. Inf. Model. 2009, 49, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Ballabio, D.; Todeschini, R. Evaluation of model predictive ability by external validation techniques. J. Chemometr. 2010, 24, 194–201. [Google Scholar] [CrossRef]
- Lin, L. A Concordance correlation-coefficient to evaluate reproducibility. Biometrics 1989, 45, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Lin, L. Assay validation using the concordance correlation-coefficient. Biometrics 1992, 48, 599–604. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Das, R.N.; Ambure, P.; Aher, R.B. Be aware of error measures. Further studies on validation of predictive QSAR models. Chemometr. Intell. Lab. 2016, 152, 18–33. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-Descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, G.W.; Jurs, P.C. QSAR and k-nearest neighbor classification analysis of selective cyclooxygenase-2 inhibitors using topologically-based numerical descriptors. J. Chem. Inf. Comput. Sci. 2001, 41, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Cox, D. The regression analysis of binary sequences. J. R. Stat. Soc. 1958, 2, 215–242. [Google Scholar]
- Walker, S.H.; Duncan, D.B. Estimation of the probability of an event as a function of several independent variables. Biometrika 1967, 54, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Sun, H. A Naive Bayes classifier for prediction of multidrug resistance reversal activity on the basis of atom typing. J. Med. Chem. 2005, 48, 4031–4039. [Google Scholar] [CrossRef] [PubMed]
- Parhizgar, H.; Dehghani, M.R.; Khazaei, A.; Dalirian, M. Application of neural networks in the prediction of surface tensions of binary mixtures. Ind. Eng. Chem. Res. 2012, 51, 2775–2781. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Lin, C. LIBSVM: A library for support vector machines. ACM. Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Plewczynski, D.; Spieser, S.; Koch, U. Assessing different classification methods for virtual screening. J. Chem. Inf. Model. 2006, 46, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Garrido, A.; Helguera, A.M.; Borges, F.; Cordeiro, M.N.D.S.; Rivero, V.; Escudero, A.G. Two new parameters based on distances in a receiver operating characteristic chart for the selection of classification models. J. Chem. Inf. Model. 2011, 51, 2746–2759. [Google Scholar] [CrossRef] [PubMed]
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.F.; Vind, C.; Brockhoff, P.B.; Refsgaard, H.H.F. In silico prediction of cytochrome P450 2D6 and 3A4 inhibition using gaussian kernel weightedk-nearest neighbor and extended connectivity fingerprints, including structural fragment analysis of inhibitors versus noninhibitors. J. Med. Chem. 2007, 50, 501–511. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Model No. | Number of Descriptors | Descriptors | R2 | R2adj | RMSEtr | CCCtr | F | Q2loo | RMSEcv | CCCcv | Q2lmo | R2Yscr | Q2Yscr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 21 | 8 | nR06 MATS6p MATS4i JGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] HOMO | 0.8071 | 0.7796 | 0.2661 | 0.8933 | 29.2961 | 0.7533 | 0.3010 | 0.8651 | 0.7379 | 0.1247 | −0.1890 |
| 2 | 27 | 8 | nR06 MATS6p MATS4i JGI4 SpMin7_Bh(i) P_VSA_MR_1 B01[C-O] F04[C-O] | 0.8033 | 0.7752 | 0.2688 | 0.8909 | 28.5870 | 0.7432 | 0.3071 | 0.8596 | 0.7267 | 0.1247 | −0.1856 |
| 3 | 29 | 8 | D/Dtr06 MATS6p MATS4i JGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] HOMO | 0.8023 | 0.7740 | 0.2695 | 0.8903 | 28.3984 | 0.7504 | 0.3028 | 0.8632 | 0.7335 | 0.1268 | −0.1880 |
| 4 | 33 | 7 | MATS6p MATS4i GATS1m JGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] | 0.7872 | 0.7611 | 0.2796 | 0.8809 | 30.1220 | 0.7322 | 0.3136 | 0.8520 | 0.7169 | 0.1097 | −0.1644 |
| 5 | 34 | 7 | Mp MATS6p MATS4i JGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] | 0.7848 | 0.7584 | 0.2811 | 0.8794 | 29.6947 | 0.7262 | 0.3171 | 0.8484 | 0.7076 | 0.1100 | −0.1636 |
| 6 | 36 | 7 | MATS6p MATS4i JGI4 SpMin7_Bh(i) H-046 B01[C-O] F04[C-O] | 0.7807 | 0.7538 | 0.2838 | 0.8768 | 28.9864 | 0.7276 | 0.3163 | 0.8491 | 0.7140 | 0.1077 | −0.1700 |
| 7 | 37 | 7 | ZM1Mad MATS6p MATS4i GGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] | 0.7797 | 0.7527 | 0.2844 | 0.8762 | 28.8222 | 0.7214 | 0.3199 | 0.8446 | 0.7045 | 0.1094 | −0.1669 |
| 8 | 38 | 7 | MATS6p MATS4i JGI4 SpMin5_Bh(s) P_VSA_MR_1 B01[C-O] F04[C-O] | 0.7786 | 0.7514 | 0.2851 | 0.8755 | 28.6378 | 0.7223 | 0.3194 | 0.8463 | 0.7040 | 0.1104 | −0.1621 |
| No. | Model No. | Number of Descriptors | Descriptors | R2ext | RMSEext | Q2F1 | Q2F2 | Q2F3 | CCCext | k | k’ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 21 | 8 | nR06 MATS6p MATS4i JGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] HOMO | 0.5401 (0.7195) | 0.3544 (0.2847) | 0.5147 (0.7041) | 0.5144 (0.7032) | 0.6581 (0.7794) | 0.7023 (0.8062) | 0.9774 (0.9957) | 1.0132 (0.9977) |
| 2 | 27 | 8 | nR06 MATS6p MATS4i JGI4 SpMin7_Bh(i) P_VSA_MR_1 B01[C-O] F04[C-O] | 0.5100 (0.6534) | 0.3709 (0.3080) | 0.4685 (0.6538) | 0.4681 (0.6527) | 0.6255 (0.7418) | 0.7003 (0.7934) | 0.9784 (0.9944) | 1.0110 (0.9994) |
| 3 | 29 | 8 | D/Dtr06 MATS6p MATS4i JGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] HOMO | 0.5153 (0.7175) | 0.3659 (0.2908) | 0.4862 (0.6912) | 0.4823 (0.6902) | 0.6355 (0.7697) | 0.6767 (0.7914) | 0.9742 (0.9933) | 1.0159 (0.9999) |
| 4 | 33 | 7 | MATS6p MATS4i GATS1m JGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] | 0.4632 (0.5963) | 0.3806 (0.3354) | 0.4381 (0.5894) | 0.4399 (0.5881) | 0.6056 (0.6938) | 0.6398 (0.7221) | 0.9787 (0.9946) | 1.0101 (0.9962) |
| 5 | 34 | 7 | Mp MATS6p MATS4i JGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] | 0.4712 (0.6587) | 0.3813 (0.3116) | 0.4381 (0.6455) | 0.4377 (0.6443) | 0.6041 (0.7356) | 0.6508 (0.7622) | 0.9752 (0.9944) | 1.0138 (0.9977) |
| 6 | 36 | 7 | MATS6p MATS4i JGI4 SpMin7_Bh(i) H-046 B01[C-O] F04[C-O] | 0.4726 | 0.3780 | 0.4478 | 0.4474 | 0.6109 | 0.6609 | 0.9804 | 1.0084 |
| 7 | 37 | 7 | ZM1Mad MATS6p MATS4i GGI4 SpMin7_Bh(i) B01[C-O] F04[C-O] | 0.6322 | 0.3295 | 0.5805 | 0.5802 | 0.7044 | 0.7851 | 0.9751 | 1.0171 |
| 8 | 38 | 7 | MATS6p MATS4i JGI4 SpMin5_Bh(s) P_VSA_MR_1 B01[C-O] F04[C-O] | 0.4710 (0.5055) | 0.3786 (0.3721) | 0.4461 (0.4944) | 0.4457 (0.4927) | 0.6097 (0.6229) | 0.6702 (0.6971) | 0.9855 (0.9941) | 1.0030 (0.9946) |
| Descriptor | Type | Chemical Meaning |
|---|---|---|
| nR06 | Ring descriptors | Number of 6-membered rings |
| MATS6p | 2D autocorrelations | Moran autocorrelation of lag 6 weighted by polarizability |
| MATS4i | 2D autocorrelations | Moran autocorrelation of lag 4 weighted by ionization potential |
| JGI4 | 2D autocorrelations | Mean topological charge index of order 4 |
| SpMin7_Bh(i) | Burden eigenvalues | Smallest eigenvalue n. 7 of Burden matrix weighted by ionization potential |
| B01[C-O] | 2D Atom Pairs | Presence/absence of C–O at topological distance 1 |
| F04[C-O] | 2D Atom Pairs | Frequency of C–O at topological distance 4 |
| EHOMO | QM descriptors | Highest occupied molecular orbital energy |
| Data Set | Model | CA | SE | SP | AUC | TP | TN | FP | FN |
|---|---|---|---|---|---|---|---|---|---|
| Training set | MACCS-ANN | 0.821 | 0.67 | 0.88 | 0.905 | 10 | 36 | 5 | 5 |
| PubChem-ANN | 0.839 | 0.73 | 0.88 | 0.885 | 11 | 36 | 5 | 4 | |
| SubFP-ANN | 0.732 | 0.33 | 0.88 | 0.872 | 5 | 36 | 5 | 10 | |
| PubChem-LR | 0.804 | 0.47 | 0.93 | 0.860 | 7 | 38 | 3 | 8 | |
| PubChem-RF | 0.804 | 0.47 | 0.93 | 0.857 | 7 | 38 | 3 | 8 | |
| Est-ANN | 0.732 | 0.33 | 0.88 | 0.840 | 5 | 36 | 5 | 10 | |
| MACCS-LR | 0.768 | 0.40 | 0.90 | 0.832 | 6 | 37 | 4 | 9 | |
| MACCS-SVM | 0.750 | 0.47 | 0.85 | 0.770 | 7 | 35 | 6 | 8 | |
| Test set | MACCS-ANN | 0.792 | 0.29 | 1.00 | 0.992 | 2 | 17 | 0 | 5 |
| PubChem-ANN | 0.708 | 0.29 | 0.88 | 0.765 | 2 | 15 | 2 | 5 | |
| SubFP-ANN | 0.667 | 0.29 | 0.82 | 0.626 | 2 | 14 | 3 | 5 | |
| PubChem-LR | 0.792 | 0.43 | 0.94 | 0.889 | 3 | 16 | 1 | 4 | |
| PubChem-RF | 0.708 | 0.14 | 0.94 | 0.693 | 1 | 16 | 1 | 6 | |
| Est-ANN | 0.750 | 0.14 | 1.00 | 0.790 | 1 | 17 | 0 | 6 | |
| MACCS-LR | 0.875 | 0.57 | 1.00 | 0.899 | 4 | 17 | 0 | 3 | |
| MACCS-SVM | 0.875 | 0.71 | 0.94 | 0.958 | 5 | 16 | 1 | 2 |
| No. | Description | SMARTS | General Structures | Representative Compounds | IG | FH |
|---|---|---|---|---|---|---|
| SubFP133 | Nitrile | [NX1]#[CX2] |  |  | 0.048 | 3.64 |
| SubFP181 | Hetero N nonbasic | [nX2,nX3+] |  |  | 0.037 | 2.73 |
| SubFP184 | Heteroaromatic | [a;!c] |  |  | 0.037 | 2.73 |
| SubFP8 | Alkylchloride | [ClX1][CX4] |  |  | 0.024 | 3.64 |
| SubFP26 | Tertiary aliph amine | [NX3H0+0,NX4H1+;!$([N][!C]);!$([N]*~[#7,#8,#15,#16])] |  |  | 0.024 | 3.64 |
| No. | Name | CAS No. | LD50 mg/kg | Log (LD50)−1 | Predicted log(LD50)−1 |
|---|---|---|---|---|---|
| 1 | Diallylnitrosamine a | 16338-97-9 | 800 (L) b | 3.10 | 3.20 |
| 2 | Dipentylnitrosamine | 13256-06-9 | 1750 (L) | 2.76 | 2.72 |
| 3 | N-Methyl-N,4-dinitrosoaniline | 99-80-9 | 1370 (L) | 2.86 | 3.20 |
| 4 | Nitroso-N-methyl-N-(2-phenyl) ethylamine | 13256-11-6 | 48 (H) b | 4.32 | 4.30 |
| 5 | N-Nitroso(2,2,2-trifluoroethyl)ethylamine a | 82018-90-4 | 960 (L) | 3.02 | 3.20 |
| 6 | Nitrosodibutylamine | 924-16-3 | 1200 (L) b | 2.92 | 3.17 |
| 7 | N-Nitrosodipropylamine | 621-64-7 | 480 (L) b | 3.32 | 3.25 |
| 8 | Nitrosoethylmethylamine | 10595-95-6 | 90 (H) b | 4.05 | 4.34 |
| 9 | 2-Nitrosomethylaminopyridine a | 16219-98-0 | 60 (H) | 4.22 | 4.23 |
| 10 | Nitrosomethylaniline | 614-00-6 | 225 (L) | 3.65 | 4.15 |
| 11 | Diisopropylnitrosamine | 601-77-4 | 850 (L) | 3.07 | 2.68 |
| 12 | N-Nitrosobis(2,2,2-trifluoro ethyl)amine | 625-89-8 | 300 (L) | 3.52 | 3.46 |
| 13 | N-Ethyl-N-tert-butylnitrosamine | 3398-69-4 | 1600 (L) b | 2.80 | 2.71 |
| 14 | N-Nitrosomethylaminoacetonitrile | 3684-97-7 | 45 (H) | 4.35 | 4.25 |
| 15 | N-Butyl-N-(4-hydroxybutyl) nitro samine | 3817-11-6 | 1800 (L) b | 2.74 | 2.34 |
| 16 | N-Nitrosomethylvinylamine | 4549-40-0 | 24 (H) | 4.62 | 4.51 |
| 17 | N-Nitroso-N-methylallylamine | 4549-43-3 | 340 (L) | 3.47 | 3.55 |
| 18 | N-Ethyl-N-butylnitrosamine | 4549-44-4 | 380 (L) b | 3.42 | 3.71 |
| 19 | N-Nitrosodibenzylamine | 5336-53-8 | 900 (L) | 3.05 | 2.92 |
| 20 | N-Nitroso-N-methylcyclohexylamine a | 5432-28-0 | 30 (H) b | 4.52 | 3.92 |
| 21 | Nitrosomethyl-n-butylamine | 7068-83-9 | 130 (H) | 3.89 | 3.99 |
| 22 | N-Ethyl-N-hydroxyethylnitrosamine | 13147-25-6 | 7500 (L) | 2.12 | 2.53 |
| 23 | N-Amyl-N-methylnitrosamine | 13256-07-0 | 120 (H) | 3.92 | 3.85 |
| 24 | Dinitrosodimethylethylenediamine | 13256-12-7 | 125 (H) b | 3.90 | 3.90 |
| 25 | Vinylethylnitrosamine | 13256-13-8 | 88 (H) | 4.06 | 3.68 |
| 26 | N-Nitrososarcosine | 13256-22-9 | 5000 (L) | 2.30 | 2.68 |
| 27 | 2-Chloro-N-methyl-N-nitrosoethanamine | 16339-16-5 | 22 (H) | 4.66 | 4.10 |
| 28 | N-Methyl(methoxymethyl)nitrosamine | 39885-14-8 | 700 (L) | 3.15 | 3.34 |
| 29 | Methyl(acetoxymethyl)nitrosamine a | 56856-83-8 | 130 (H) | 3.89 | 3.83 |
| 30 | Acetoxymethylbutylnitrosamine a | 56986-36-8 | 1500 (L) | 2.82 | 2.89 |
| 31 | 1-Methoxy-ethyl-ethylnitrosamine | 61738-03-2 | 1000 (L) b | 3.00 | 2.84 |
| 32 | Methoxymethyl-ethylnitrosamine | 61738-04-3 | 540 (L) | 3.27 | 3.12 |
| 33 | 1-Methoxy-ethyl-methylnitrosamine | 61738-05-4 | 240 (L) | 3.62 | 3.35 |
| 34 | Acetoxymethylpropylnitrosamine | 66017-91-2 | 1000 (L) | 3.00 | 3.05 |
| 35 | Methyl(butyroxymethyl)nitrosamine | 67557-56-6 | 800 (L) b | 3.10 | 3.20 |
| 36 | Acetoxymethyltrideuteromethylnitrosamine | 67557-57-7 | 120 (H) | 3.92 | 3.88 |
| 37 | N-Nitroso-N-phenylhydroxylamine | 148-97-0 | 490 (L) b | 3.31 | 3.53 |
| 38 | N-methyl-n-benzylnitrosamine | 937-40-6 | 18 (H) b | 4.74 | 4.22 |
| 39 | 4-(Methylnitrosoamino)benzaldehyde a | 7431-19-8 | 2000 (L) | 2.70 | 2.76 |
| 40 | 3-(N-Nitrosomethylamino)sulfolan | 13256-21-8 | 750 (L) | 3.12 | 2.88 |
| 41 | Aethyl-4-picolylnitrosamin | 13256-23-0 | 40 (H) | 4.40 | 4.02 |
| 42 | N,N’-Dimethylnitrosourea | 13256-32-1 | 280 (L) | 3.55 | 3.50 |
| 43 | N-Nitrososarcosine ethyl ester | 13344-50-8 | 4000 (L) | 2.40 | 2.65 |
| 44 | 4-Nitrosomethylaminopyridine | 16219-99-1 | 200 (L) | 3.70 | 3.81 |
| 45 | N-Nitrosoethylisopropylamine | 16339-04-1 | 1100 (L) b | 2.96 | 3.19 |
| 46 | N-Nitrosotrimethylhydrazine | 16339-14-3 | 95 (H) b | 4.02 | 4.05 |
| 47 | N-Nitrosodiacetonitrile | 16339-18-7 | 163 (H) | 3.79 | 3.89 |
| 48 | N-Nitroso-N-ethylbenzylamine | 20689-96-7 | 250 (L) b | 3.60 | 3.66 |
| 49 | N-Nitroso-O,N-diethylhydroxylamine | 56235-95-1 | 1000 (L) b | 3.00 | 2.79 |
| 50 | N-Nitroso-N-(2-methylbenzyl)methylamine | 62783-48-6 | 90 (H) | 4.05 | 3.96 |
| 51 | N-Methyl-N-nitroso-(3-methylphenyl)methylamine | 62783-49-7 | 600 (L) | 3.22 | 3.41 |
| 52 | N-Methyl-N-nitroso-(4-methylphenyl)methylamine | 62783-50-0 | 400 (L) b | 3.40 | 3.89 |
| 53 c | N-Nitroso-N-methyl-1(1-phenyl)-ethylamine a | 68690-89-1 | 600 (L) | 3.22 | 4.00 |
| 54 | N-Nitroso-N-methyl-2-(2-phenyl)-propylamine | 68690-90-4 | 2100 (L) | 2.68 | 2.82 |
| 55 | 3-Nitrosomethylaminopyridine | 69658-91-9 | 10 (H) | 5.00 | 4.40 |
| 56 | N-Nitrosodiethylamine a | 55-18-5 | 220 (L) | 3.66 | 3.62 |
| 57 | N-Nitrosodimethylamine | 62-75-9 | 37 (H) | 4.43 | 4.53 |
| 58 | N-Nitrosodiphenylamine | 86-30-6 | 1825 (L) | 2.74 | 2.74 |
| 59 | N-Nitroso-3,6-dihydro-1,2-oxazine | 3276-41-3 | 900 (L) | 3.05 | 3.05 |
| 60 | R(−)-N-Nitroso-2-methylpiperidine | 14026-03-0 | 600 (L) | 3.22 | 2.94 |
| 61 | S(+)-N-Nitroso-2-methylpiperidine | 36702-44-0 | 600 (L) | 3.22 | 3.00 |
| 62 | N-Nitrosoheptamethyleneimine a | 20917-49-1 | 283 (L) | 3.55 | 3.58 |
| 63 | N-Nitrosomorpholine | 59-89-2 | 282 (L) | 3.55 | 3.23 |
| 64 | N-Nitrosopyrrolidine | 930-55-2 | 900 (L) | 3.05 | 3.35 |
| 65 | 1-Nitrosopiperazine | 5632-47-3 | 2260 (L) | 2.65 | 3.39 |
| 66 | N-Nitrosopiperidine | 100-75-4 | 200 (L) | 3.70 | 3.39 |
| 67 | N-Nitroso-tetrahydro-1,2-oxazine | 40548-68-3 | 830 (L) b | 3.08 | 2.95 |
| 68 | N-Nitrosoperhydroazepine a | 932-83-2 | 336 (L) | 3.47 | 3.51 |
| 69 | N-Nitrosoindoline | 7633-57-0 | 320 (L) | 3.49 | 3.40 |
| 70 | N-Nitroso-N’-methylpiperazine a | 16339-07-4 | 100 (H) b | 4.00 | 3.51 |
| 71 | N-Nitrosoazacyclononane | 20917-50-4 | 566 (L) b | 3.25 | 3.40 |
| 72 | 3-Nitrosotetrahydro-1,3-oxazine | 35627-29-3 | 600 (L) | 3.22 | 3.29 |
| 73 | N-Nitroso-1,3-oxazolidine | 39884-52-1 | 1500 (L) | 2.82 | 2.92 |
| 74 | 1-Amyl-1-nitrosourea a | 10589-74-9 | 560 (L) | 3.25 | 3.30 |
| 75 | N-Nitroso-N-butylurea | 869-01-2 | 400 (L) b | 3.40 | 3.49 |
| 76 | N-Nitroso-N-ethylurea | 759-73-9 | 300 (L) | 3.52 | 3.46 |
| 77 | N-Nitroso-N-methylurea | 684-93-5 | 110 (H) | 3.96 | 4.27 |
| 78 | Propylnitrosourea | 816-57-9 | 480 (L) | 3.32 | 3.13 |
| 79 | N-Nitroso-N-methylbiuret a | 13860-69-0 | 450 (L) b | 3.35 | 3.73 |
| 80 | Ethylnitrosobiuret a | 32976-88-8 | 1050 (L) | 2.98 | 3.53 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, T.; Sun, G.; Zhao, L.; Cui, X.; Zhong, R. QSAR and Classification Study on Prediction of Acute Oral Toxicity of N-Nitroso Compounds. Int. J. Mol. Sci. 2018, 19, 3015. https://doi.org/10.3390/ijms19103015
Fan T, Sun G, Zhao L, Cui X, Zhong R. QSAR and Classification Study on Prediction of Acute Oral Toxicity of N-Nitroso Compounds. International Journal of Molecular Sciences. 2018; 19(10):3015. https://doi.org/10.3390/ijms19103015
Chicago/Turabian StyleFan, Tengjiao, Guohui Sun, Lijiao Zhao, Xin Cui, and Rugang Zhong. 2018. "QSAR and Classification Study on Prediction of Acute Oral Toxicity of N-Nitroso Compounds" International Journal of Molecular Sciences 19, no. 10: 3015. https://doi.org/10.3390/ijms19103015
APA StyleFan, T., Sun, G., Zhao, L., Cui, X., & Zhong, R. (2018). QSAR and Classification Study on Prediction of Acute Oral Toxicity of N-Nitroso Compounds. International Journal of Molecular Sciences, 19(10), 3015. https://doi.org/10.3390/ijms19103015