Prostaglandin D2 Induces Ca2+ Sensitization of Contraction without Affecting Cytosolic Ca2+ Level in Bronchial Smooth Muscle


Abstract
:1. Introduction
2. Results
2.1. Effects of Prostaglandin D2 (PGD2) on Bronchial Smooth Muscle (BSM) Function
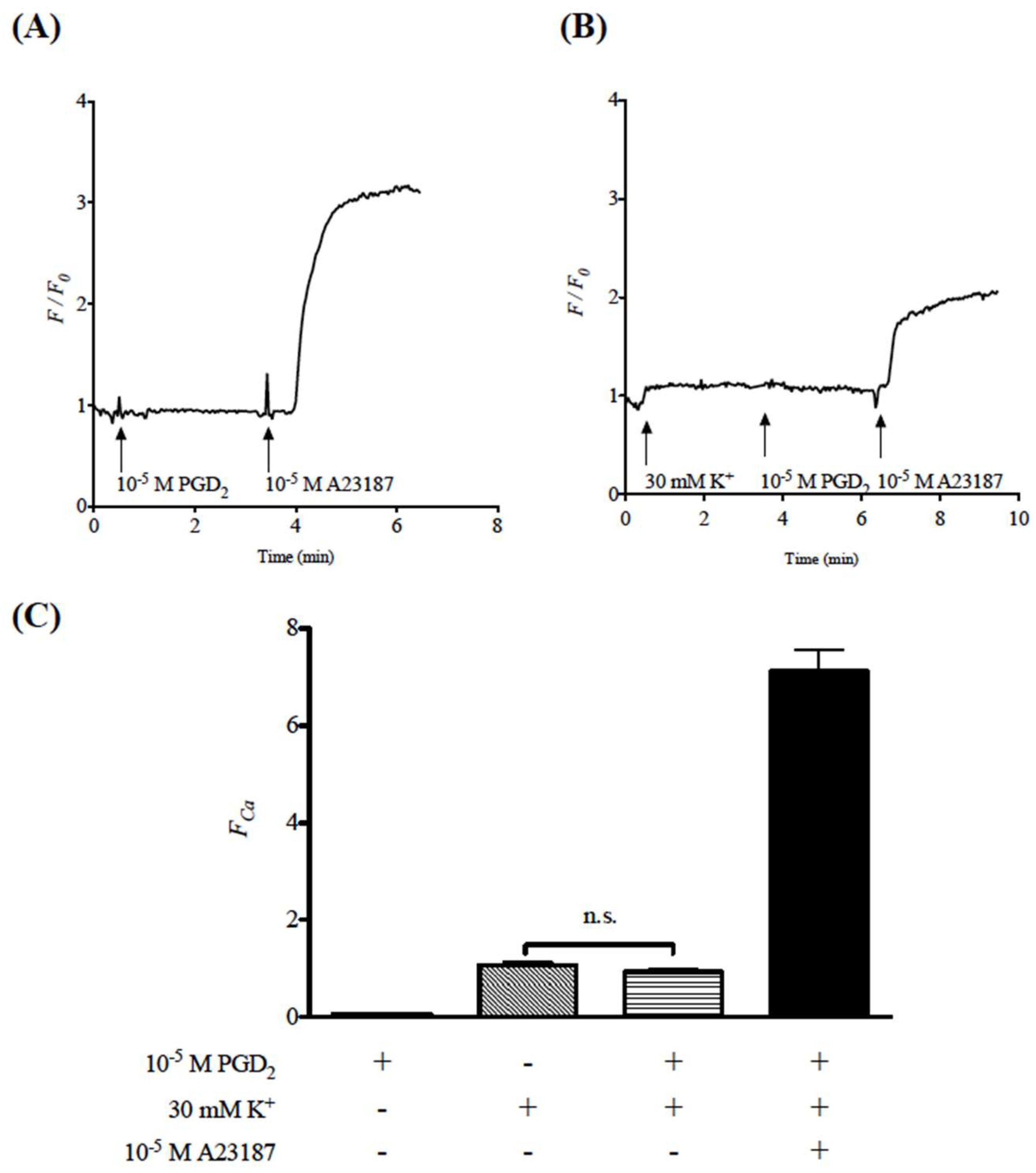
2.2. Effects of Prostaglandin D2 (PGD2) on Cytosolic Ca2+ Level in Human Bronchial Smooth Muscle Cells (hBSMCs)
2.3. Activation of RhoA/Rho-Kinase Signaling by Prostaglandin D2 (PGD2)
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Pharmacological Reagents
4.3. Determination of Bronchial Smooth Muscle (BSM) Responsiveness
4.4. Determination of Active Form of RhoA in BSM
4.5. Cell Culture and [Ca2+]cyto Measurement
4.6. RT-PCR Analyses
4.7. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACh | acetylcholine |
| AHR | airway hyperresponsiveness |
| ANOVA | analysis of variance |
| BAL | bronchoalveolar lavage |
| BSM | bronchial smooth muscle |
| COX | cyclooxygenase |
| CRTH2 | chemoattractant receptor-homologous molecule on Th2 cells |
| ERK | extracellular signal-regulated kinase |
| FCa | normalized ratios of the Ca2+ fluorescence intensities |
| F/F0 | ratio of the Ca2+ fluorescence intensity to that at time 0 (baseline) |
| Fluo-8/AM | Fluo-8 acetoxymethyl ester |
| GPCR | G protein-coupled receptor |
| GST | glutathione S-transferase |
| GTP | guanosine triphosphate |
| PG | prostaglandin |
| SDS | sodium dodecyl sulfate |
| TXA2 | thromboxane A2 |
References
- Björck, T.; Gustafsson, L.E.; Dahlén, S.E. Isolated bronchi from asthmatics are hyperresponsive to adenosine, which apparently acts indirectly by liberation of leukotrienes and histamine. Am. Rev. Respir. Dis. 1992, 145, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Seow, C.Y.; Schellenberg, R.R.; Paré, P.D. Structural and functional changes in the airway smooth muscle of asthmatic subjects. Am. J. Respir. Crit. Care Med. 1998, 158, S178–S186. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.G.; Duguet, A.; Eidelman, D.H. The contribution of airway smooth muscle to airway narrowing and airway hyperresponsiveness in disease. Eur. Respir. J. 2000, 16, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cheng, Z.; Kong, H.; Wang, Y.; Unruh, H.; Stephens, N.L.; Laviolette, M. Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L1181–L1189. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.D. Critical role of actin-associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respir. Res. 2015, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Dworski, R.; Sheller, J.R.; Wickersham, N.E.; Oates, J.A.; Brigham, K.L.; Roberts, L.J.; Fitzgerald, G.A. Allergen-stimulated release of mediators into sheep bronchoalveolar lavage fluid. Effect of cyclooxygenase inhibition. Am. Rev. Respir. Dis. 1989, 139, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.K.; Zhang, Z.; Ray, R.; Choi, M.S.; Chowdhury, B.; Pattabiraman, N.; Mukherjee, A.B. Uteroglobin represses allergen-induced inflammatory response by blocking PGD2 receptor-mediated functions. J. Exp. Med. 2004, 199, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Chilton, F.H.; Averill, F.J.; Hubbard, W.C.; Fonteh, A.N.; Triggiani, M.; Liu, M.C. Antigen-induced generation of lyso-phospholipids in human airways. J. Exp. Med. 1996, 183, 2235–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartert, T.V.; Dworski, R.T.; Mellen, B.G.; Oates, J.A.; Murray, J.J.; Sheller, J.R. Prostaglandin E2 decreases allergen-stimulated release of prostaglandin D2 in airways of subjects with asthma. Am. J. Respir. Crit. Care Med. 2000, 162, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Hirata, M.; Tanaka, H.; Takahashi, Y.; Murata, T.; Kabashima, K.; Sugimoto, Y.; Kobayashi, T.; Ushikubi, F.; Aze, Y.; et al. Prostaglandin D2 as a mediator of allergic asthma. Science 2000, 287, 2013–2017. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Gyles, S.L.; Wettey, F.R.; Gazi, L.; Townsend, E.; Hunter, M.G.; Pettipher, R. Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J. Immunol. 2005, 175, 6531–6536. [Google Scholar] [CrossRef] [PubMed]
- Underwood, D.C.; Muccitelli, R.M.; Luttmann, M.A.; Hay, D.W.; Torphy, T.J.; Wasserman, M.A. Differential antagonism of airway contractile responses to prostaglandin (PG)D2 and 9 α, 11 β-PGF2 by atropine, SK&F 88046 and SQ 29,548 in the guinea pig. J. Pharmacol. Exp. Ther. 1994, 268, 304–310. [Google Scholar] [PubMed]
- Tamaoki, J.; Sekizawa, K.; Graf, P.D.; Nadel, J.A. Cholinergic neuromodulation by prostaglandin D2 in canine airway smooth muscle. J. Appl. Physiol. 1987, 63, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- Sturino, C.F.; O’Neill, G.; Lachance, N.; Boyd, M.; Berthelette, C.; Labelle, M.; Li, L.; Roy, B.; Scheigetz, J.; Tsou, N.; et al. Discovery of a potent and selective prostaglandin D2 receptor antagonist, [(3R)-4-(4-chloro-benzyl)-7-fluoro-5-(methylsulfonyl)-1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl]-acetic acid (MK-0524). J. Med. Chem. 2007, 50, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.A.; Bradley, M.E.; Riddy, D.M.; Willard, E.; Reilly, J.; Miah, A.; Bauer, C.; Watson, S.J.; Sandham, D.A.; Dubois, G.; et al. Fevipiprant (QAW039), a slowly dissociating CRTh2 antagonist with the potential for improved clinical efficacy. Mol. Pharmacol. 2016, 89, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, J.J.; Botello-Smith, W.M.; Luo, Y. Probing the gating mechanism of the mechanosensitive channel Piezo1 with the small molecule Yoda1. Nat. Commun. 2018, 9, 2029. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Ueno, A.; Shinozaki, K.; Takeyama, H.; Nakazawa, S.; Sakai, H.; Misawa, M. Involvement of RhoA-mediated Ca2+ sensitization in antigen-induced bronchial smooth muscle hyperresponsiveness in mice. Respir. Res. 2005, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerthoffer, W.T.; Solway, J.; Camoretti-Mercado, B. Emerging targets for novel therapy of asthma. Curr. Opin. Pharmacol. 2013, 13, 324–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, Y.; Danno, S.; Suto, R.; Suto, W.; Yamane, Y.; Hanazaki, M.; Katayama, H.; Sakai, H. Intranasal administration of recombinant progranulin inhibits bronchial smooth muscle hyperresponsiveness in mouse allergic asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L215–L223. [Google Scholar] [CrossRef] [PubMed]
- Modena, B.D.; Dazy, K.; White, A.A. Emerging concepts: Mast cell involvement in allergic diseases. Transl. Res. 2016, 174, 98–121. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Sheldrick, R.L. Prostanoid-induced contraction of human bronchial smooth muscle is mediated by TP-receptors. Br. J. Pharmacol. 1989, 96, 688–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraki, A.; Kume, H.; Oguma, T.; Makino, Y.; Ito, S.; Shimokata, K.; Honjo, H.; Kamiya, K. Role of Ca2+ mobilization and Ca2+ sensitization in 8-iso-PGF2α-induced contraction in airway smooth muscle. Clin. Exp. Allergy 2009, 39, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Sakai, H.; Suenaga, H.; Kamata, K.; Misawa, M. Enhanced Ca2+ sensitization of the bronchial smooth muscle contraction in antigen-induced airway hyperresponsive rats. Res. Commun. Mol. Pathol. Pharmacol. 1999, 106, 77–85. [Google Scholar] [PubMed]
- Chiba, Y.; Takada, Y.; Miyamoto, S.; MitsuiSaito, M.; Karaki, H.; Misawa, M. Augmented acetylcholine-induced, Rho-mediated Ca2+ sensitization of bronchial smooth muscle contraction in antigen-induced airway hyperresponsive rats. Br. J. Pharmacol. 1999, 127, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Cheah, E.Y.; Mann, T.S.; Burcham, P.C.; Henry, P.J. Influenza A infection attenuates relaxation responses of mouse tracheal smooth muscle evoked by acrolein. Biochem. Pharmacol. 2015, 93, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Donovan, C.; Bailey, S.R.; Tran, J.; Haitsma, G.; Ibrahim, Z.A.; Foster, S.R.; Tang, M.L.; Royce, S.G.; Bourke, J.E. Rosiglitazone elicits in vitro relaxation in airways and precision cut lung slices from a mouse model of chronic allergic airways disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1219–L1228. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.J.; Deshpande, D.A.; Tiegs, B.C.; Misior, A.M.; Yan, H.; Hershfeld, A.V.; Rich, T.C.; Panettieri, R.A.; An, S.S.; Penn, R.B. Beta-Agonist-mediated relaxation of airway smooth muscle is protein kinase A-dependent. J. Biol. Chem. 2014, 289, 23065–23074. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Lee, S.N.; Aoyagi, H.; Yamasaki, Y.; Yoo, J.Y.; Park, B.; Shin, D.M.; Yoon, H.G.; Yoon, J.H. The extracellular signal-regulated kinase mitogen-activated protein kinase/ribosomal S6 protein kinase 1 cascade phosphorylates cAMP response element-binding protein to induce MUC5B gene expression via D-prostanoid receptor signaling. J. Biol. Chem. 2011, 286, 34199–34214. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | RefSeq Accession | Sequence | Amplicon Size | |
|---|---|---|---|---|
| human PTGDR | NM_000953 | Sense | 5′-TCTGCGCGCTACCTTTCATG-3′ | 85 bp |
| Antisense | 5′-TCCTCGTGGACCATCTGGATA-3′ | |||
| human PTGDR2 | NM_004778 | Sense | 5′-CCTCTGTGCCCAGAGCCCCACGATGTCGGC-3′ | 114 bp |
| Antisense | 5′-ATGTAGCGGATGCTGGTGTTG-3′ | |||
| human PTGER1 | NM_000955 | Sense | 5′-GATGGTGGGCCAGCTTGTC-3′ | 72 bp |
| Antisense | 5′-GCCACCAACACCAGCATTG-3′ | |||
| human PTGER2 | NM_000956 | Sense | 5′-GTGCTGACAAGGCACTTCATGT-3′ | 87 bp |
| Antisense | 5′-TGTTCCTCCAAAGGCCAAGTAC-3′ | |||
| human PTGER3 | NM_198714 | Sense | 5′-AAGGCCACGGCATCTCAGT-3′ | 76 bp |
| Antisense | 5′-TGATCCCCATAAGCTGAATGG-3′ | |||
| human PTGER4 | NM_000958 | Sense | 5′-CTTGGAGGCAGGAATTTGCTT-3′ | 77 bp |
| Antisense | 5′-AAAGTCCTCAGTGAGGTGGTGTCT-3′ | |||
| human PTGFR | NM_000959 | Sense | 5′-GCACATTGATGGGCAACTAGAA-3′ | 91 bp |
| Antisense | 5′-GCACCTATCATTGGCATGTAGCT-3′ | |||
| human PTGIR | NM_000960 | Sense | 5′-GCCGATCAGCTGCTGTTTCT-3′ | 75 bp |
| Antisense | 5′-TTTCCTCTGTCCCTCACTCTCTTC-3′ | |||
| human TBXA2R | NM_001060 | Sense | 5′-ACGGAGAAGGAGCTGCTCATC-3′ | 84 bp |
| Antisense | 5′-GCGGCGGAACAGGATATACA-3′ | |||
| human GAPDH | NM_002046 | Sense | 5′-GGAGCCAAAAGGGTCATCATCTC-3′ | 282 bp |
| Antisense | 5′-AGGGATGATGTTCTGGAGAGCC-3′ |
| Gene Name | RefSeq Accession | Sequence | Amplicon Size | |
|---|---|---|---|---|
| mouse Ptgdr1 | NM_008962 | Sense | 5′-CAACCTGGGTGCCATGTAC-3′ | 112 bp |
| Antisense | 5′-GGACCCGTGCCTGTAGTCT-3′ | |||
| mouse Ptgdr2 | NM_009962 | Sense | 5′-CTGCACCTGGCGCTATC-3′ | 174 bp |
| Antisense | 5′-GTCCAGGCTAATGGCACT-3′ | |||
| mouse Ptger1 | NM_013641 | Sense | 5′-TACATGGGATGCTCGAAACA-3′ | 223 bp |
| Antisense | 5′-TTTTAGGCCGTGTGGGTAG-3′ | |||
| mouse Ptger2 | NM_008964 | Sense | 5′-ATGCACCTGCTGCTTATCGT-3′ | 196 bp |
| Antisense | 5′-TAATGGCCAGGAGAATGAGG-3′ | |||
| mouse Ptger3 | NM_001359745 | Sense | 5′-TGCTGGCTCTGGTGGTGAC-3′ | 258 bp |
| Antisense | 5′-ACTCCTTCTCCTTTCCCATCTGTG-3′ | |||
| mouse Ptger4 | NM_001136079 | Sense | 5′-CCATCGCCACATACATGAAG-3′ | 209 bp |
| Antisense | 5′-TGCACAGATGGCGAAGAGTG-3′ | |||
| mouse Ptgfr | NM_008966 | Sense | 5′-CTGCTCCGGACACAACCACTC-3′ | 191 bp |
| Antisense | 5′-GGTTCTCCGTCTGGCAGGTTG-3′ | |||
| mouse Ptgir | NM_008967 | Sense | 5′-GGATGAAGTTTACCACCTGATTCTGC-3′ | 196 bp |
| Antisense | 5′-AGCCTTTCGGAAAAGGATGAAGAC-3′ | |||
| mouse Tbxa2r | NM_009325 | Sense | 5′-TTTCGCCCGGTGAACATC-3′ | 255 bp |
| Antisense | 5′-GGCTCGCCAGTCCAACAA-3′ | |||
| mouse Gapdh | NM_001289726 | Sense | 5′-CCTCGTCCCGTAGACAAAATG-3′ | 100 bp |
| Antisense | 5′-TCTCCACTTTGCCACTGCAA-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suto, W.; Ando, Y.; Hirabayashi, T.; Takenoya, F.; Shioda, S.; Kamei, J.; Sakai, H.; Chiba, Y. Prostaglandin D2 Induces Ca2+ Sensitization of Contraction without Affecting Cytosolic Ca2+ Level in Bronchial Smooth Muscle. Int. J. Mol. Sci. 2018, 19, 3036. https://doi.org/10.3390/ijms19103036
Suto W, Ando Y, Hirabayashi T, Takenoya F, Shioda S, Kamei J, Sakai H, Chiba Y. Prostaglandin D2 Induces Ca2+ Sensitization of Contraction without Affecting Cytosolic Ca2+ Level in Bronchial Smooth Muscle. International Journal of Molecular Sciences. 2018; 19(10):3036. https://doi.org/10.3390/ijms19103036
Chicago/Turabian StyleSuto, Wataru, Yusuke Ando, Takahiro Hirabayashi, Fumiko Takenoya, Seiji Shioda, Junzo Kamei, Hiroyasu Sakai, and Yoshihiko Chiba. 2018. "Prostaglandin D2 Induces Ca2+ Sensitization of Contraction without Affecting Cytosolic Ca2+ Level in Bronchial Smooth Muscle" International Journal of Molecular Sciences 19, no. 10: 3036. https://doi.org/10.3390/ijms19103036
APA StyleSuto, W., Ando, Y., Hirabayashi, T., Takenoya, F., Shioda, S., Kamei, J., Sakai, H., & Chiba, Y. (2018). Prostaglandin D2 Induces Ca2+ Sensitization of Contraction without Affecting Cytosolic Ca2+ Level in Bronchial Smooth Muscle. International Journal of Molecular Sciences, 19(10), 3036. https://doi.org/10.3390/ijms19103036