Comprehensive Analysis of MicroRNA–Messenger RNA from White Yak Testis Reveals the Differentially Expressed Molecules Involved in Development and Reproduction

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
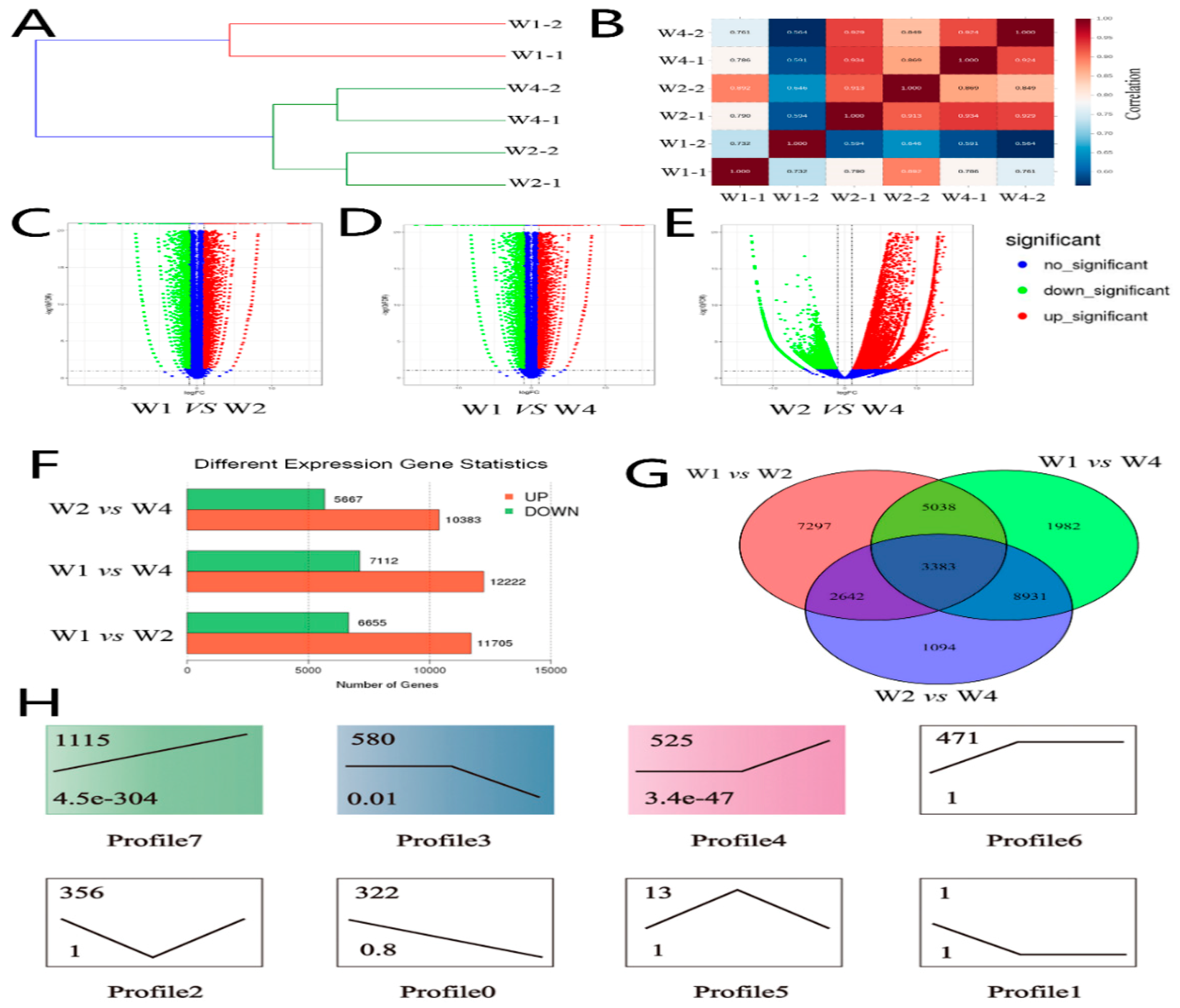
2.1. Identifying Differentially Expressed Genes of Transcriptome Sequencing
2.2. Identifying Differentially Expressed MicroRNA of Small RNA Sequencing
2.3. Gene Ontology (GO) and Pathways Analysis of Different Expressed Target Genes
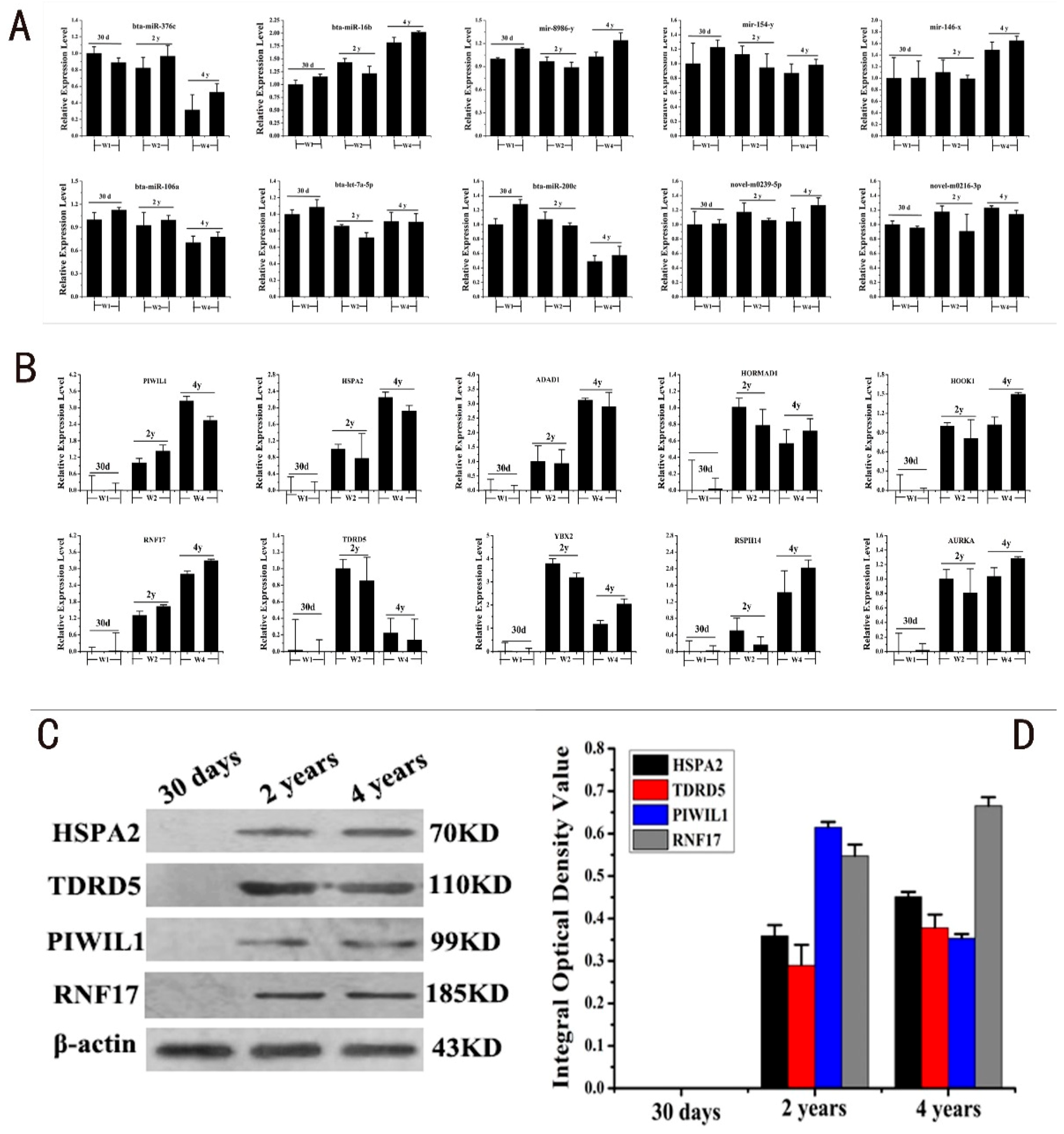
2.4. RT-PCR and Western Blot Validation of DERs and DEGs
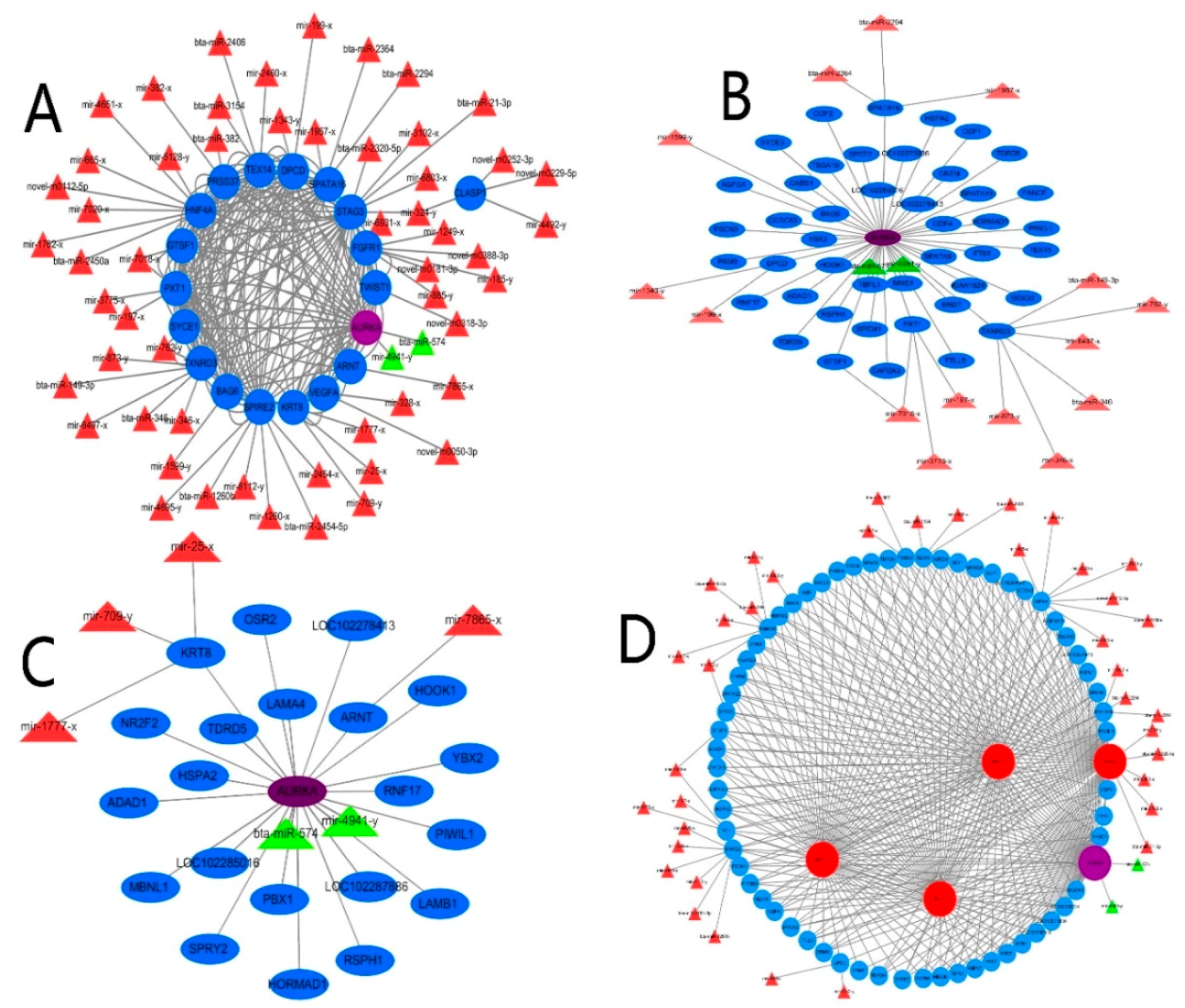
2.5. Integrated Network Analysis of Differentially Expressed MicroRNA–Messenger RNA in Testis Development, Reproduction and Spermatogenesis
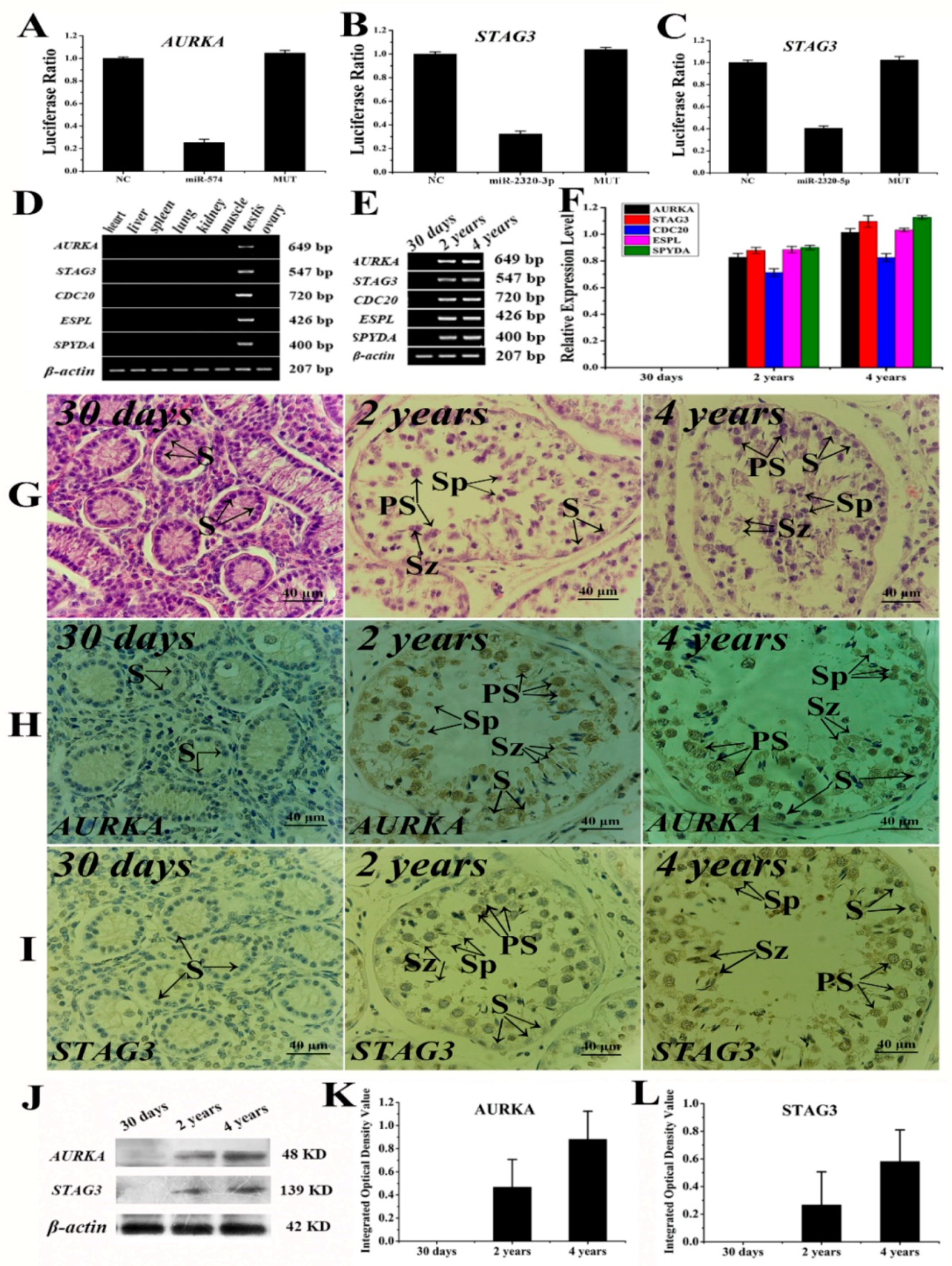
2.6. The Verification Analysis of Core DERs and DEGs of Yak Testis in Development and Reproduction
3. Discussion
4. Materials and Methods
4.1. Samples Preparation and Collection
4.2. Total RNA and MicroRNA Preparation and Sequencing
4.3. RNA and MicroRNAs Analysis
4.4. Functional Enrichment and Cluster Analysis
4.5. PCR Assays for Target Genes and MicroRNAs
4.6. Western Blot, Immunohistochemistry and Hematoxylin-Eosin Staining Assays
4.7. Luciferase Reporter Assays for Key MicroRNAs and Target Genes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DER | Differentially Expressed microRNA |
| DEG | Differentially Expressed gene |
| FPKM | Fragments Per Kilobase of transcript per Million mapped reads |
| TPM | Transcripts Per Million |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PCR | Polymerase Chain Reaction |
| UTR | Untranslated Regions |
References
- Zhang, X.; Wang, K.; Wang, L.; Yang, Y.; Ni, Z.; Xie, X.; Shao, X.; Han, J.; Wan, D.; Qiu, Q. Genome-wide patterns of copy number variation in the chinese yak genome. BMC Genom. 2016, 17, 379. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Xu, S.; Hu, L.; Zhao, N.; Liu, Z.; Ma, L.; Liu, H.; Zhao, X. Effect of dietary types on feed intakes, growth performance and economic benefit in tibetan sheep and yaks on the qinghai-tibet plateau during cold season. PLoS ONE 2017, 12, e0169187. [Google Scholar] [CrossRef] [PubMed]
- Zi, X.D. Reproduction in female yaks (Bos grunniens) and opportunities for improvement. Theriogenology 2003, 59, 1303–1312. [Google Scholar] [CrossRef]
- Long, R.J.; Dong, S.K.; Wei, X.H.; Pu, X.P. The effect of supplementary feeds on the bodyweight of yaks in cold season. Livest. Prod. Sci. 2005, 93, 197–204. [Google Scholar] [CrossRef]
- Ou, Y.; Dores, C.; Rodriguez-Sosa, J.R. Primary cilia in the developing pig testis. Cell Tissue Res. 2014, 358, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, M.D.; Nef, D.S. MicroRNAs in the testis: Building up male fertility. J. Androl. 2010, 31, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chen, Y.; Zhang, H.; Chen, Y.; Shen, X.; Shi, C.; Liu, Y.; Yuan, W. Integrated microRNA-mRNA analyses reveal opll specific microRNA regulatory network using high-throughput sequencing. Sci. Rep. 2016, 6, 21580. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.D.; Huang, H.Y.; Chou, C.H.; Sun, Y.M.; Hsu, M.T.; Tsou, A.P. Integrated analyses to reconstruct microRNA-mediated regulatory networks in mouse liver using high-throughput profiling. BMC Genom. 2015, 16, S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Chen, L.; Zhang, X.; Liu, X.; Chen, Y.; Mo, D. An integrated analysis revealed different microRNA-mRNA profiles during skeletal muscle development between landrace and lantang pigs. Sci. Rep. 2017, 7, 2516. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, B.; Geng, J.; Zhou, J.; Zheng, R.; Jin, C.; Li, F.; Peng, J.; Jiang, S. Integrated analysis of miRNA/mRNA network in placenta identifies key factors associated with labor onset of large white and qingping sows. Sci. Rep. 2015, 5, 13074. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.G.; Calin, G.A.; Meloon, B.; Gamliel, N.; Sevignani, C.; Ferracin, M.; Dumitru, C.D.; Shimizu, M.; Zupo, S.; Dono, M. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc. Natl. Acad. Sci. USA 2004, 101, 9740–9744. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Takizawa, T.; Luo, S.S.; Ishibashi, O.; Kawahigashi, Y.; Mizuguchi, Y.; Ishikawa, T.; Mori, M.; Kanda, T.; Goto, T. MicroRNA (miRNA) cloning analysis reveals sex differences in miRNA expression profiles between adult mouse testis and ovary. Reproduction 2008, 136, 811. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ju, Z.; Li, Q.; Hou, Q.; Wang, C.; Li, J.; Li, R.; Wang, L.; Tao, S.; Hang, S. Solexa sequencing of novel and differentially expressed microRNAs in testicular and ovarian tissues in holstein cattle. Int. J. Biol. Sci. 2011, 7, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bao, J.; Kim, M.; Yuan, S.; Tang, C.; Zheng, H.; Mastick, G.S.; Xu, C.; Yan, W. Two miRNA clusters, mir-34b/c and mir-449, are essential for normal brain development, motile ciliogenesis, and spermatogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, E2851. [Google Scholar] [CrossRef] [PubMed]
- Schurch, N.J.; Schofield, P.; Gierliå„Ski, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA Publ. RNA Soc. 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, D.R. Searching the mouse genome informatics (MGI) resources for information on mouse biology from genotype to phenotype. Curr. Protoc. Bioinform. 2004. [Google Scholar] [CrossRef]
- Zhang, M.; Dai, X.; Sun, Y.; Lu, Y.; Zhou, C.; Miao, Y.; Wang, Y.; Xiong, B. Stag3 regulates microtubule stability to maintain euploidy during mouse oocyte meiotic maturation. Oncotarget 2016, 8, 1593. [Google Scholar] [CrossRef] [PubMed]
- Yabuta, Y.; Ohta, H.; Abe, T.; Kurimoto, K.; Chuma, S.; Saitou, M. Tdrd5 is required for retrotransposon silencing, chromatoid body assembly, and spermiogenesis in mice. J. Cell Biol. 2011, 192, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Goodheart, M.; Chuma, S.; Nakatsuji, N.; Page, D.C.; Wang, P.J. Rnf17, a component of the mammalian germ cell nuage, is essential for spermiogenesis. Development 2005, 132, 4029. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Dong, Y.; Liu, W.; Ma, X.; Shi, R.; Chen, H.; Cui, Z.; Ao, L.; Zhang, H.; Cao, J. Epigenetic regulation of sox30 is associated with testis development in mice. PLoS ONE 2014, 9, e97203. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Mukherjee, A.; Wu, J.; Zhang, L.; Teves, M.E.; Li, H.; Nambiar, S.; Henderson, S.C.; Horwitz, A.R.; Iii, J.F.S. Sperm associated antigen 6 (spag6) regulates fibroblast cell growth, morphology, migration and ciliogenesis. Sci. Rep. 2015, 5, 16506. [Google Scholar] [CrossRef] [PubMed]
- Svingen, T.; Koopman, P. Building the mammalian testis: Origins, differentiation, and assembly of the component cell populations. Genes Dev. 2013, 27, 2409–2426. [Google Scholar] [CrossRef] [PubMed]
- Skaftnesmo, K.O.; Edvardsen, R.B.; Furmanek, T.; Crespo, D.; Andersson, E.; Kleppe, L.; Taranger, G.L.; Bogerd, J.; Schulz, R.W.; Wargelius, A. Integrative testis transcriptome analysis reveals differentially expressed miRNAs and their mRNA targets during early puberty in atlantic salmon. BMC Genom. 2017, 18, 801. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhong, H.; Zhou, Y.; Yu, F.; Gao, Y.; Luo, Y.; Tang, Z.; Guo, Z.; Guo, E.; Gan, X. Identification and characterization of microRNAs in ovary and testis of nile tilapia (oreochromis niloticus) by using solexa sequencing technology. PLoS ONE 2014, 9, e86821. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Guo, Q.; Chang, G.; Qiu, L.; Liu, X.; Bi, Y.; Zhang, Y.; Wang, H.; Lu, W.; Ren, L. Discovery of microRNAs during early spermatogenesis in chicken. PLoS ONE 2017, 12, e0177098. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wu, S.; Zhao, W.; Mipam, T.; Liu, J.; Liu, W.; Yi, C.; Shah, M.A.; Yu, S.; Cai, X. Differentially expressed microRNAs between cattleyak and yak testis. Sci. Rep. 2018, 8, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiener, G.; Han, J.L.; Long, R.J.; Wiener, G.; Han, J.L.; Long, R.J. The yak. Rap Publ. 2011, 44, 57–58. [Google Scholar]
- Wiener, G.; Han, J.; Long, R. The Yak; FAO Regional Office for Asia and the Pacific: Bangkok, Thailand, 2003; pp. 57–58. [Google Scholar]
- Bourc’His, D.; Bestor, T.H. Origins of extreme sexual dimorphism in genomic imprinting. Cytogenet. Genome Res. 2006, 113, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Mruk, D.D.; Cheng, C.Y. The mammalian blood-testis barrier: Its biology and regulation. Endocr. Rev. 2015, 36, 564. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, F.; Zhang, X.; Li, L.; Wang, L.; Shi, B.; Xu, J. Depression of hspa2 in human testis is associated with spermatogenic impairment and fertilization rate in icsi treatment for azoospermic individuals. J. Assisted Reprod. Genet. 2014, 31, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, B.; Yu, H. The bub1-plk1 kinase complex promotes spindle checkpoint signalling through CDC20 phosphorylation. Nat. Commun. 2016, 7, 10818. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Hamada, M.; Malureanu, L.; Jeganathan, K.B.; Zhou, W.; Morbeck, D.E.; van Deursen, J.M. CDC20 is critical for meiosis I and fertility of female mice. PLoS Genet. 2010, 6, e1001147. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Lopes, S.M.C.D.S.; Kaneda, M.; Tang, F.; Hajkova, P.; Lao, K.; O’Carroll, D.; Das, P.P.; Tarakhovsky, A.; Miska, E.A. MicroRNA biogenesis is required for mouse primordial germ cell development and spermatogenesis. PLoS ONE 2008, 3, e1738. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Gupta, M.K.; Ji, Y.S.; Sang, J.U.; Lee, H.T. MicroRNA signature in testes-derived male germ-line stem cells. Mol. Hum. Reprod. 2010, 16, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Jiang, J.; Kokkinaki, M.; Tang, L.; Zeng, W.; Gallicano, I.; Dobrinski, I.; Dym, M. miRNA-20 and miRNA-106a regulate spermatogonial stem cell renewal at the post-transcriptional level via targeting STAT3 and ccnd1. Stem Cells 2013, 31, 2205–2217. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Lu, X.U.; Chang, G.; Guo, Q.; Liu, X.; Yulin, B.I.; Yu, Z.; Wang, H.; Wang, K.; Wei, L.U. DNA methylation-mediated transcription factors regulatepiwil1expression during chicken spermatogenesis. J. Reprod. Dev. 2016, 62, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.P. The cycle of the seminiferous epithelium in humans: A need to revisit? J. Androl. 2008, 29, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Nihi, F.; Gomes, M.L.; Carvalho, F.A.; Reis, A.B.; Martello, R.; Melo, R.C.; Almeida, F.R.; Chiarinigarcia, H. Revisiting the human seminiferous epithelium cycle. Hum. Reprod. 2017, 32, 1170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, J.Y.; Chen, Y.N.; Yuan, F.; Zhang, H.; Yan, F.H.; Wang, M.J.; Wang, G.; Su, M.; Lu, G. Whole genome and transcriptome sequencing of matched primary and peritoneal metastatic gastric carcinoma. Sci. Rep. 2015, 5, 13750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. Tophat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat. Protoc. 2012, 7, 562. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. Li b, dewey cn. Rsem: Accurate transcript quantification from RNA-seq data with or without a reference genome. Bmc bioinformatics 12:323. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-seq. Bioinformatics 2011, 27, 2325. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.; Adam, R.; Loyal, G.; Geo, P.; Daehwan, K.; David, R.K.; Harold, P.; Steven, L.S.; John, L.R.; Lior, P. Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat. Protoc. 2014, 7, 562. [Google Scholar]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, X.; Zhu, J.; Gu, Y.; Zhao, W.; Zou, J.; Guo, Z. Go-function: Deriving biologically relevant functions from statistically significant functions. Brief. Bioinform. 2012, 13, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. Kegg for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Yang, Y.; Wang, Z.; Zhao, S.; Mu, Y.; Li, K. Integrated analysis of miRNA and mRNA paired expression profiling of prenatal skeletal muscle development in three genotype pigs. Sci. Rep. 2015, 5, 15544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, S.; Bhattacharyya, M. A biologically inspired measure for coexpression analysis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 929. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Chiang, V.L. Facile means for quantifying microRNA expression by real-time pcr. Biotechniques 2005, 39, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gong, J.; Wang, X.; Wu, X.; Li, Y.; Ma, Y.; Zhang, Y.; Zhao, X. Molecular cloning, bioinformatics analysis and expression of insulin-like growth factor 2 from tianzhu white yak, bos grunniens. Int. J. Mol. Sci. 2014, 15, 504–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Q.; Gong, J.; Du, J.; Zhang, Y.; Zhao, X. Yak igf2 promotes fibroblast proliferation via suppression of igf1r and pi3kcg expression. Genes 2018, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhang, Q.; Wang, Q.; Ma, Y.; Du, J.; Zhang, Y.; Zhao, X. Identification and verification of potential piRNAs from domesticated yak testis. Reproduction 2018, 155, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Otali, D.; Fredenburgh, J.; Oelschlager, D.K.; Grizzle, W.E. A standard tissue as a control for histochemical and immunohistochemical staining. Biotech. Histochem. 2016, 91, 309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Y.; Ma, Y.; Zhao, X. Molecular characteristics of the ho1 gene in yak are potentially adaptive for high altitude habitats. J. Comput. Theor. Nanosci. 2017, 14, 2698–2705. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Wang, Q.; Zhang, Y.; Cheng, S.; Hu, J.; Ma, Y.; Zhao, X. Comprehensive Analysis of MicroRNA–Messenger RNA from White Yak Testis Reveals the Differentially Expressed Molecules Involved in Development and Reproduction. Int. J. Mol. Sci. 2018, 19, 3083. https://doi.org/10.3390/ijms19103083
Zhang Q, Wang Q, Zhang Y, Cheng S, Hu J, Ma Y, Zhao X. Comprehensive Analysis of MicroRNA–Messenger RNA from White Yak Testis Reveals the Differentially Expressed Molecules Involved in Development and Reproduction. International Journal of Molecular Sciences. 2018; 19(10):3083. https://doi.org/10.3390/ijms19103083
Chicago/Turabian StyleZhang, Quanwei, Qi Wang, Yong Zhang, Shuru Cheng, Junjie Hu, Youji Ma, and Xingxu Zhao. 2018. "Comprehensive Analysis of MicroRNA–Messenger RNA from White Yak Testis Reveals the Differentially Expressed Molecules Involved in Development and Reproduction" International Journal of Molecular Sciences 19, no. 10: 3083. https://doi.org/10.3390/ijms19103083
APA StyleZhang, Q., Wang, Q., Zhang, Y., Cheng, S., Hu, J., Ma, Y., & Zhao, X. (2018). Comprehensive Analysis of MicroRNA–Messenger RNA from White Yak Testis Reveals the Differentially Expressed Molecules Involved in Development and Reproduction. International Journal of Molecular Sciences, 19(10), 3083. https://doi.org/10.3390/ijms19103083