Biochemical Characterization of the GBA2 c.1780G>C Missense Mutation in Lymphoblastoid Cells from Patients with Spastic Ataxia



Abstract
:1. Introduction
2. Results
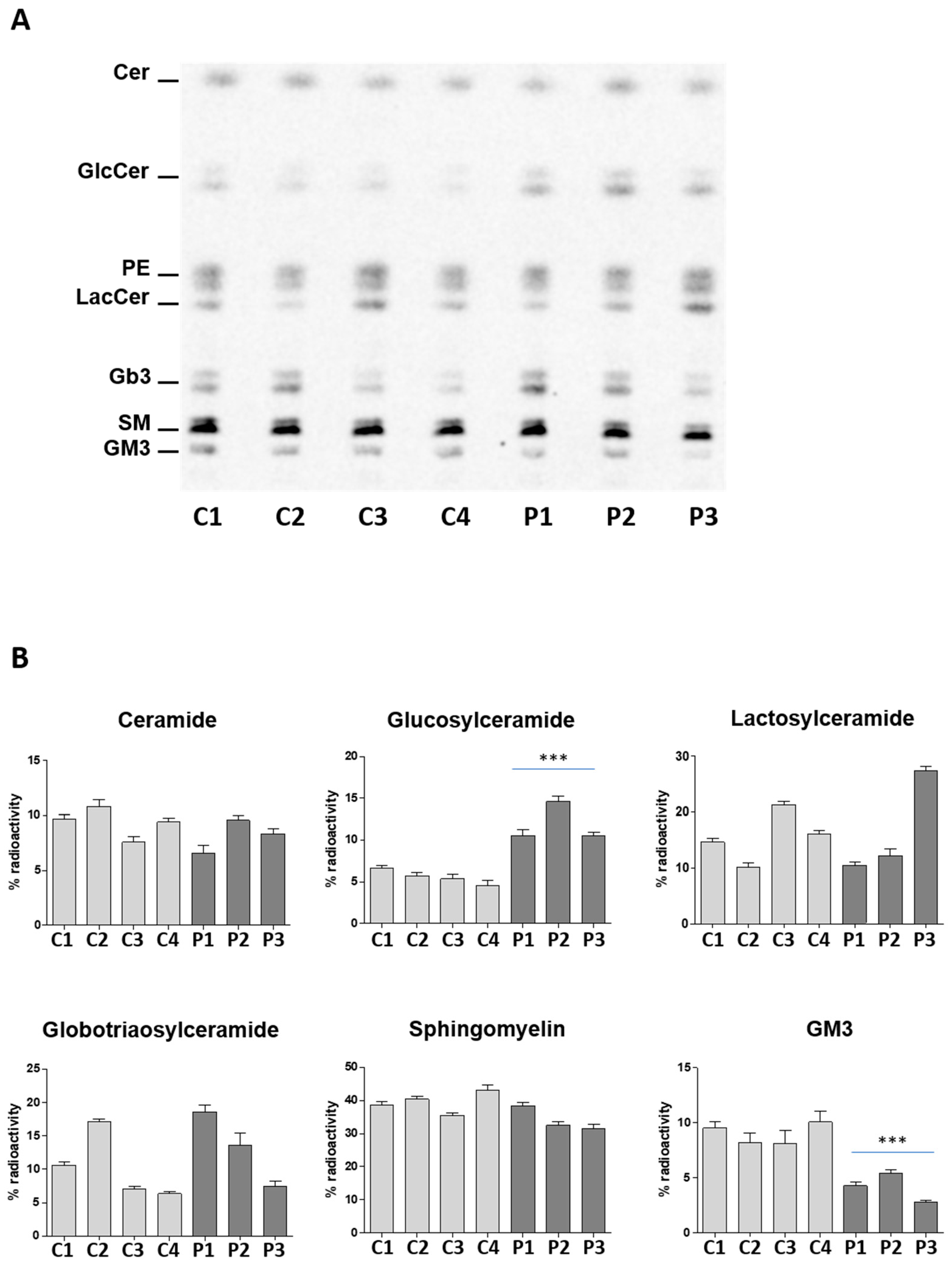
2.1. The c.1780G>C Mutation Results in NLGase Loss of Activity and GlcCer Accumulation
2.2. GCase Activity is Up-Regulated in GBA2-Deficient LCLs Particularly at the PM
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Evaluation of Enzymatic Activities in Cell Lysates
4.3. Evaluation of Enzymatic Activities at the Cell Surface of Living Cells
4.4. Real-Time PCR
4.5. Immunoblotting
4.6. Cell Sphingolipid Labelling with [1-3H]-Sphingosine
4.7. Lipid Analysis by SFC-MS/MS
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sonnino, G. Transport processes in magnetically confined plasmas in the nonlinear regime. Chaos 2006. [Google Scholar] [CrossRef] [PubMed]
- Jeckel, D.; Karrenbauer, A.; Burger, K.N.; van Meer, G.; Wieland, F. Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. J. Cell Biol. 1992, 117, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamari, F.; Mochel, F.; Sedel, F.; Saudubray, J.M. Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: Toward a new category of inherited metabolic diseases. J. Inherit. Metab. Dis. 2013, 36, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Kuivenhoven, J.A.; Hegele, R.A. Mining the genome for lipid genes. Biochim. Biophys. Acta 2014, 1842, 1993–2009. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Loberto, N.; Chigorno, V.; Prinetti, A.; Sonnino, S. Remodeling of sphingolipids by plasma membrane associated enzymes. Neurochem. Res. 2011, 36, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Magini, A.; Polchi, A.; Urbanelli, L.; Cesselli, D.; Beltrami, A.; Tancini, B.; Emiliani, C. TFEB activation promotes the recruitment of lysosomal glycohydrolases beta-hexosaminidase and beta-galactosidase to the plasma membrane. Biochem. Biophys. Res. Commun. 2013, 440, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Kanfer, J.N.; Shapiro, D. Metabolism of Glucocerebrosides. Ii. Evidence of an Enzymatic Deficiency in Gaucher’s Disease. Biochem. Biophys. Res. Commun. 1965, 18, 221–225. [Google Scholar] [CrossRef]
- Grace, M.E.; Newman, K.M.; Scheinker, V.; Berg-Fussman, A.; Grabowski, G.A. Analysis of human acid beta-glucosidase by site-directed mutagenesis and heterologous expression. J. Biol. Chem. 1994, 269, 2283–2291. [Google Scholar] [PubMed]
- Cobucci-Ponzano, B.; Aurilia, V.; Riccio, G.; Henrissat, B.; Coutinho, P.M.; Strazzulli, A.; Padula, A.; Corsaro, M.M.; Pieretti, G.; Pocsfalvi, G.; et al. A new archaeal beta-glycosidase from Sulfolobus solfataricus: seeding a novel retaining beta-glycan-specific glycoside hydrolase family along with the human non-lysosomal glucosylceramidase GBA2. J. Biol. Chem. 2010, 285, 20691–20703. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Mesonero, J.E.; Stutz, A.; Poiree, J.C.; Giudicelli, J.; Cursio, R.; Gloor, S.M.; Semenza, G. Intestinal lactase-phlorizin hydrolase (LPH): the two catalytic sites; the role of the pancreas in pro-LPH maturation. FEBS Lett. 1998, 435, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Rempel, B.P.; Withers, S.G. Covalent inhibitors of glycosidases and their applications in biochemistry and biology. Glycobiology 2008, 18, 570–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshland, D.E., Jr.; Clarke, E. Mechanism of hydrolysis of adenosinetriphosphate catalyzed by lobster muscle. J. Biol. Chem. 1953, 205, 917–924. [Google Scholar] [PubMed]
- Matern, H.; Boermans, H.; Lottspeich, F.; Matern, S. Molecular cloning and expression of human bile acid beta-glucosidase. J. Biol. Chem. 2001, 276, 37929–37933. [Google Scholar] [CrossRef] [PubMed]
- Korschen, H.G.; Yildiz, Y.; Raju, D.N.; Schonauer, S.; Bonigk, W.; Jansen, V.; Kremmer, E.; Kaupp, U.B.; Wachten, D. The non-lysosomal beta-glucosidase GBA2 is a non-integral membrane-associated protein at the endoplasmic reticulum (ER) and Golgi. J. Biol. Chem. 2013, 288, 3381–3393. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Masilamani, A.P.; Illuzzi, G.; Loberto, N.; Scandroglio, F.; Prinetti, A.; Chigorno, V.; Sonnino, S. Activity of plasma membrane beta-galactosidase and beta-glucosidase. FEBS Lett. 2009, 583, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Loberto, N.; Bassi, R.; Ferraretto, A.; Perego, S.; Lanteri, P.; Chigorno, V.; Sonnino, S.; Prinetti, A. Plasma membrane-associated glycohydrolases activation by extracellular acidification due to proton exchangers. Neurochem. Res. 2012, 37, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, Y.; Matern, H.; Thompson, B.; Allegood, J.C.; Warren, R.L.; Ramirez, D.M.; Hammer, R.E.; Hamra, F.K.; Matern, S.; Russell, D.W. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Invest. 2006, 116, 2985–2994. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Carmona, M.A.; Sandhoff, R.; Tacke, F.; Vogt, A.; Weber, S.; Canbay, A.E.; Rogler, G.; Sauerbruch, T.; Lammert, F.; Yildiz, Y. Beta-glucosidase 2 knockout mice with increased glucosylceramide show impaired liver regeneration. Liver Int. 2012, 32, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Raju, D.; Schonauer, S.; Hamzeh, H.; Flynn, K.C.; Bradke, F.; Vom Dorp, K.; Dormann, P.; Yildiz, Y.; Trotschel, C.; Poetsch, A.; et al. Accumulation of glucosylceramide in the absence of the beta-glucosidase GBA2 alters cytoskeletal dynamics. PLoS Genet. 2015, 11, e1005063. [Google Scholar] [CrossRef] [PubMed]
- Schonauer, S.; Korschen, H.G.; Penno, A.; Rennhack, A.; Breiden, B.; Sandhoff, K.; Gutbrod, K.; Dormann, P.; Raju, D.N.; Haberkant, P.; et al. Identification of a feedback loop involving beta-glucosidase 2 and its product sphingosine sheds light on the molecular mechanisms in Gaucher disease. J. Biol. Chem. 2017, 292, 6177–6189. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Schule, R.; Smets, K.; Rastetter, A.; Boukhris, A.; Loureiro, J.L.; Gonzalez, M.A.; Mundwiller, E.; Deconinck, T.; Wessner, M.; et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 92, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Votsi, C.; Zamba-Papanicolaou, E.; Middleton, L.T.; Pantzaris, M.; Christodoulou, K. A novel GBA2 gene missense mutation in spastic ataxia. Ann. Hum. Genet. 2014, 78, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.B.; Eleuch-Fayache, G.; Schottlaender, L.V.; Nehdi, H.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Hernandez, D.G.; Sailer, A.; Liu, G.; et al. Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am. J. Hum. Genet. 2013, 92, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Citterio, A.; Arnoldi, A.; Panzeri, E.; D’Angelo, M.G.; Filosto, M.; Dilena, R.; Arrigoni, F.; Castelli, M.; Maghini, C.; Germiniasi, C.; et al. Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J. Neurol. 2014, 261, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Sultana, S.; Reichbauer, J.; Schule, R.; Mochel, F.; Synofzik, M.; van der Spoel, A.C. Lack of enzyme activity in GBA2 mutants associated with hereditary spastic paraplegia/cerebellar ataxia (SPG46). Biochem. Biophys. Res. Commun. 2015, 465, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Haugarvoll, K.; Johansson, S.; Rodriguez, C.E.; Boman, H.; Haukanes, B.I.; Bruland, O.; Roque, F.; Jonassen, I.; Blomqvist, M.; Telstad, W.; et al. GBA2 Mutations Cause a Marinesco-Sjogren-Like Syndrome: Genetic and Biochemical Studies. PLoS ONE 2017, 12, e0169309. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Bassi, R.; Prinetti, A.; Chiricozzi, E.; Pappalardi, B.; Chigorno, V.; Di Muzio, N.; Loberto, N.; Sonnino, S. Ionizing radiations increase the activity of the cell surface glycohydrolases and the plasma membrane ceramide content. Glycoconj. J. 2012. [Google Scholar] [CrossRef] [PubMed]
- Loberto, N.; Tebon, M.; Lampronti, I.; Marchetti, N.; Aureli, M.; Bassi, R.; Giri, M.G.; Bezzerri, V.; Lovato, V.; Cantu, C.; et al. GBA2-encoded beta-glucosidase activity is involved in the inflammatory response to Pseudomonas aeruginosa. PLoS ONE 2014, 9, e104763. [Google Scholar] [CrossRef] [PubMed]
- Samarani, M.; Loberto, N.; Solda, G.; Straniero, L.; Asselta, R.; Duga, S.; Lunghi, G.; Zucca, F.A.; Mauri, L.; Ciampa, M.G.; et al. A lysosome-plasma membrane-sphingolipid axis linking lysosomal storage to cell growth arrest. FASEB. J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aureli, M.; Bassi, R.; Loberto, N.; Regis, S.; Prinetti, A.; Chigorno, V.; Aerts, J.M.; Boot, R.G.; Filocamo, M.; Sonnino, S. Cell surface associated glycohydrolases in normal and Gaucher disease fibroblasts. J. Inherit. Metab. Dis. 2012, 35, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, S.; Cavalieri, C.; Magini, A.; Tancini, B.; Basso, L.; Lemansky, P.; Hasilik, A.; Li, Y.T.; Chigorno, V.; Orlacchio, A.; et al. Identification of plasma membrane associated mature beta-hexosaminidase A, active towards GM2 ganglioside, in human fibroblasts. FEBS Lett. 2005, 579, 5501–5506. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 2005, 21, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Walden, C.M.; Sandhoff, R.; Chuang, C.C.; Yildiz, Y.; Butters, T.D.; Dwek, R.A.; Platt, F.M.; van der Spoel, A.C. Accumulation of glucosylceramide in murine testis, caused by inhibition of beta-glucosidase 2: Implications for spermatogenesis. J. Biol. Chem. 2007, 282, 32655–32664. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Loberto, N.; Lanteri, P.; Chigorno, V.; Prinetti, A.; Sonnino, S. Cell surface sphingolipid glycohydrolases in neuronal differentiation and aging in culture. J. Neurochem. 2011, 116, 891–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boot, R.G.; Verhoek, M.; Donker-Koopman, W.; Strijland, A.; van Marle, J.; Overkleeft, H.S.; Wennekes, T.; Aerts, J.M. Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J. Biol. Chem. 2007, 282, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Overkleeft, H.S.; Renkema, G.H.; Neele, J.; Vianello, P.; Hung, I.O.; Strijland, A.; van der Burg, A.M.; Koomen, G.J.; Pandit, U.K.; Aerts, J.M. Generation of specific deoxynojirimycin-type inhibitors of the non-lysosomal glucosylceramidase. J. Biol. Chem. 1998, 273, 26522–26527. [Google Scholar] [CrossRef] [PubMed]
- Schiumarini, D.; Loberto, N.; Mancini, G.; Bassi, R.; Giussani, P.; Chiricozzi, E.; Samarani, M.; Munari, S.; Tamanini, A.; Cabrini, G.; et al. Evidence for the Involvement of Lipid Rafts and Plasma Membrane Sphingolipid Hydrolases in Pseudomonas aeruginosa Infection of Cystic Fibrosis Bronchial Epithelial Cells. Mediators Inflamm. 2017, 2017, 1730245. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, J.; Pierce, J.S.; Snider, J.; Rembiesa, B.; Szulc, Z.M.; Bielawska, A. Comprehensive quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods Mol. Biol. 2009, 579, 443–467. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Hexosylceramides of LCLs from Controls and Patients (Pmoles/mg Cell Proteins) | ||
|---|---|---|
| WT | c.1780 G>C | |
| Glucosylceramide | 1080 ± 107 | 2009 ± 114 |
| Galactosylceramide | 9 ± 3 | 6 ± 2 |
| Glucosylceramide Molecular Species of LCLs from Controls and Patients (Pmoles/mg Cell Proteins) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C14 | C16 | C18 | C18:1 | C20 | C22 | C22:1 | C24 | C24:1 | C26 | C26:1 | ||
| GlcCer | WT | 13 ± 2 | 611 ± 100 | 24 ± 5 | 1 ± 0.1 | 28 ± 7 | 76 ± 14 | 6 ± 1 | 143 ± 22 | 150 ± 21 | 9 ± 1 | 15 ± 2 |
| c.1780 G>C | 21 ± 1 | 1139 ± 103 | 51 ± 2 | 1.3 ± 0.2 | 63 ± 7 | 145±7 | 11 ± 1 | 261 ± 20 | 278 ± 44 | 15 ± 2 | 21 ± 1 | |
| Enzymes | Cell Lysate | PM | ||
|---|---|---|---|---|
| WT | c.1780 G>C | WT | c.1780 G>C | |
| β-Galactosidase | 1284 ± 246 | 1399 ± 149 | 14 ± 4 | 30 ± 4 |
| β-Hexosaminidase | 1868 ± 233 | 1961 ± 206 | 24 ± 6 | 35 ± 15 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malekkou, A.; Samarani, M.; Drousiotou, A.; Votsi, C.; Sonnino, S.; Pantzaris, M.; Chiricozzi, E.; Zamba-Papanicolaou, E.; Aureli, M.; Loberto, N.; et al. Biochemical Characterization of the GBA2 c.1780G>C Missense Mutation in Lymphoblastoid Cells from Patients with Spastic Ataxia. Int. J. Mol. Sci. 2018, 19, 3099. https://doi.org/10.3390/ijms19103099
Malekkou A, Samarani M, Drousiotou A, Votsi C, Sonnino S, Pantzaris M, Chiricozzi E, Zamba-Papanicolaou E, Aureli M, Loberto N, et al. Biochemical Characterization of the GBA2 c.1780G>C Missense Mutation in Lymphoblastoid Cells from Patients with Spastic Ataxia. International Journal of Molecular Sciences. 2018; 19(10):3099. https://doi.org/10.3390/ijms19103099
Chicago/Turabian StyleMalekkou, Anna, Maura Samarani, Anthi Drousiotou, Christina Votsi, Sandro Sonnino, Marios Pantzaris, Elena Chiricozzi, Eleni Zamba-Papanicolaou, Massimo Aureli, Nicoletta Loberto, and et al. 2018. "Biochemical Characterization of the GBA2 c.1780G>C Missense Mutation in Lymphoblastoid Cells from Patients with Spastic Ataxia" International Journal of Molecular Sciences 19, no. 10: 3099. https://doi.org/10.3390/ijms19103099
APA StyleMalekkou, A., Samarani, M., Drousiotou, A., Votsi, C., Sonnino, S., Pantzaris, M., Chiricozzi, E., Zamba-Papanicolaou, E., Aureli, M., Loberto, N., & Christodoulou, K. (2018). Biochemical Characterization of the GBA2 c.1780G>C Missense Mutation in Lymphoblastoid Cells from Patients with Spastic Ataxia. International Journal of Molecular Sciences, 19(10), 3099. https://doi.org/10.3390/ijms19103099