Bivalent Ligand UDCA-LPE Inhibits Pro-Fibrogenic Integrin Signalling by Inducing Lipid Raft-Mediated Internalization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. UDCA-LPE Induces Translocation of Integrins
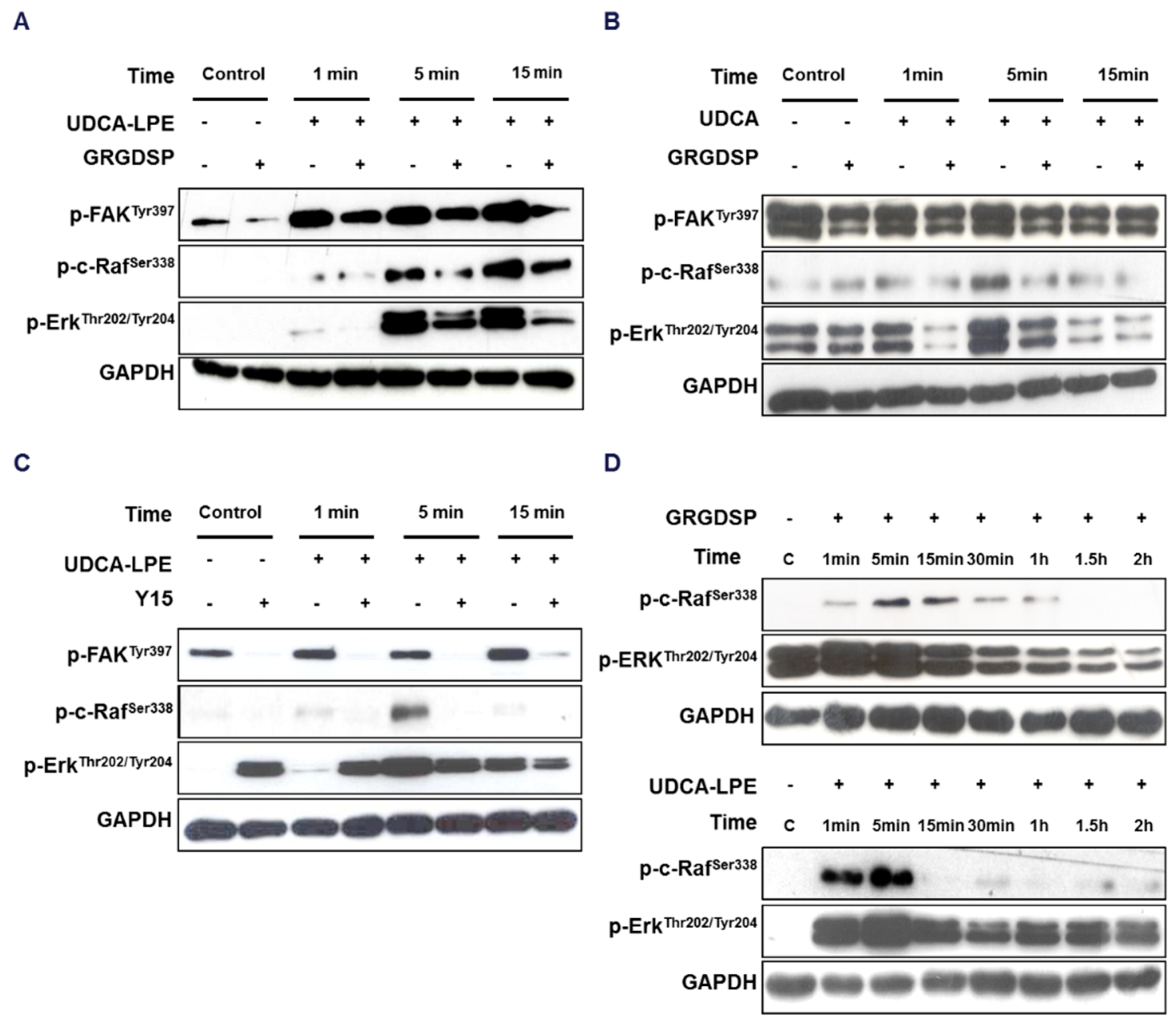
2.2. Integrin Translocation by UDCA-LPE Suppresses FAK and SRC Phosphorylation
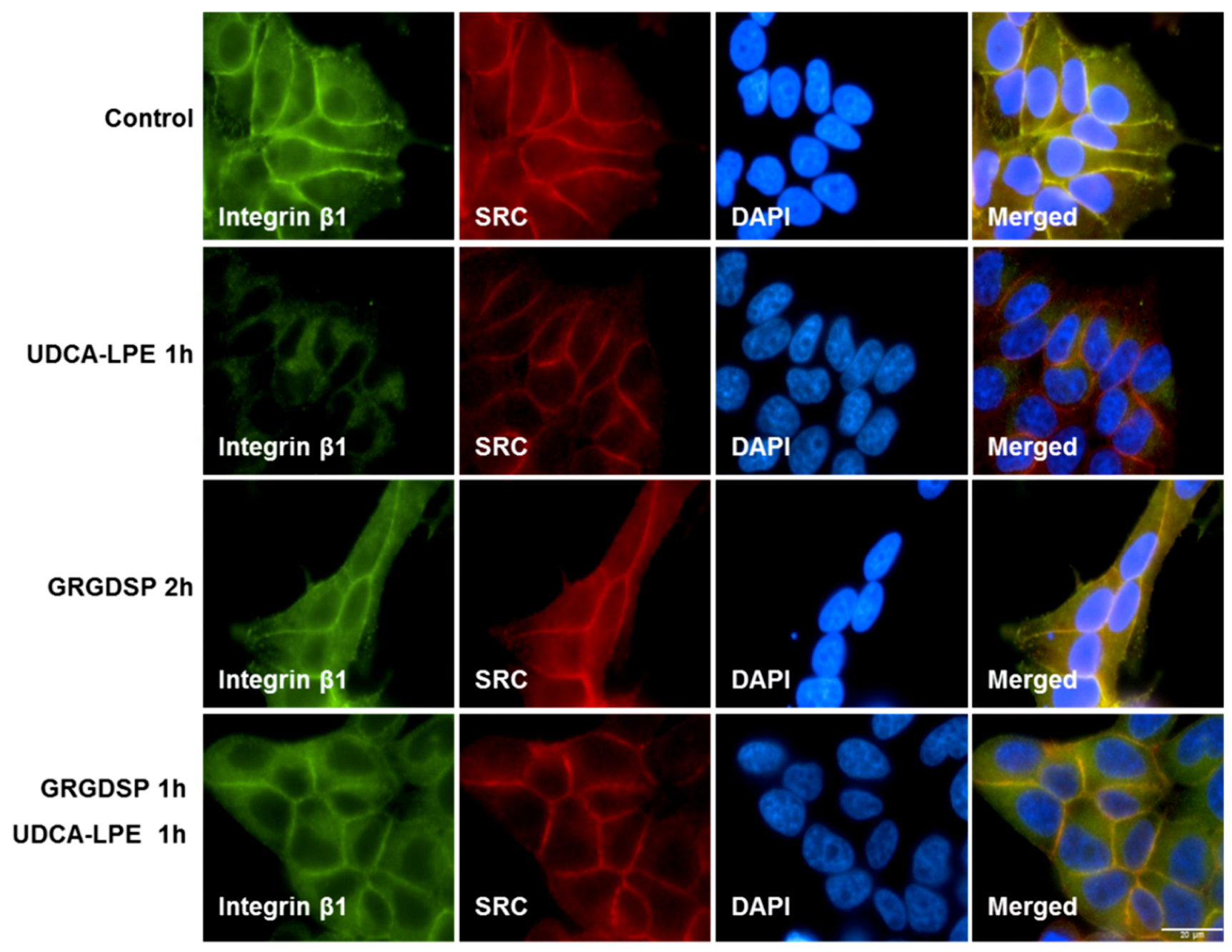
2.3. RGD-Containing Peptide GRGDSP Inhibits UDCA-LPE-Induced Translocation of Integrins
2.4. UDCA-LPE Binds to Integrins with Its UDCA-Moiety
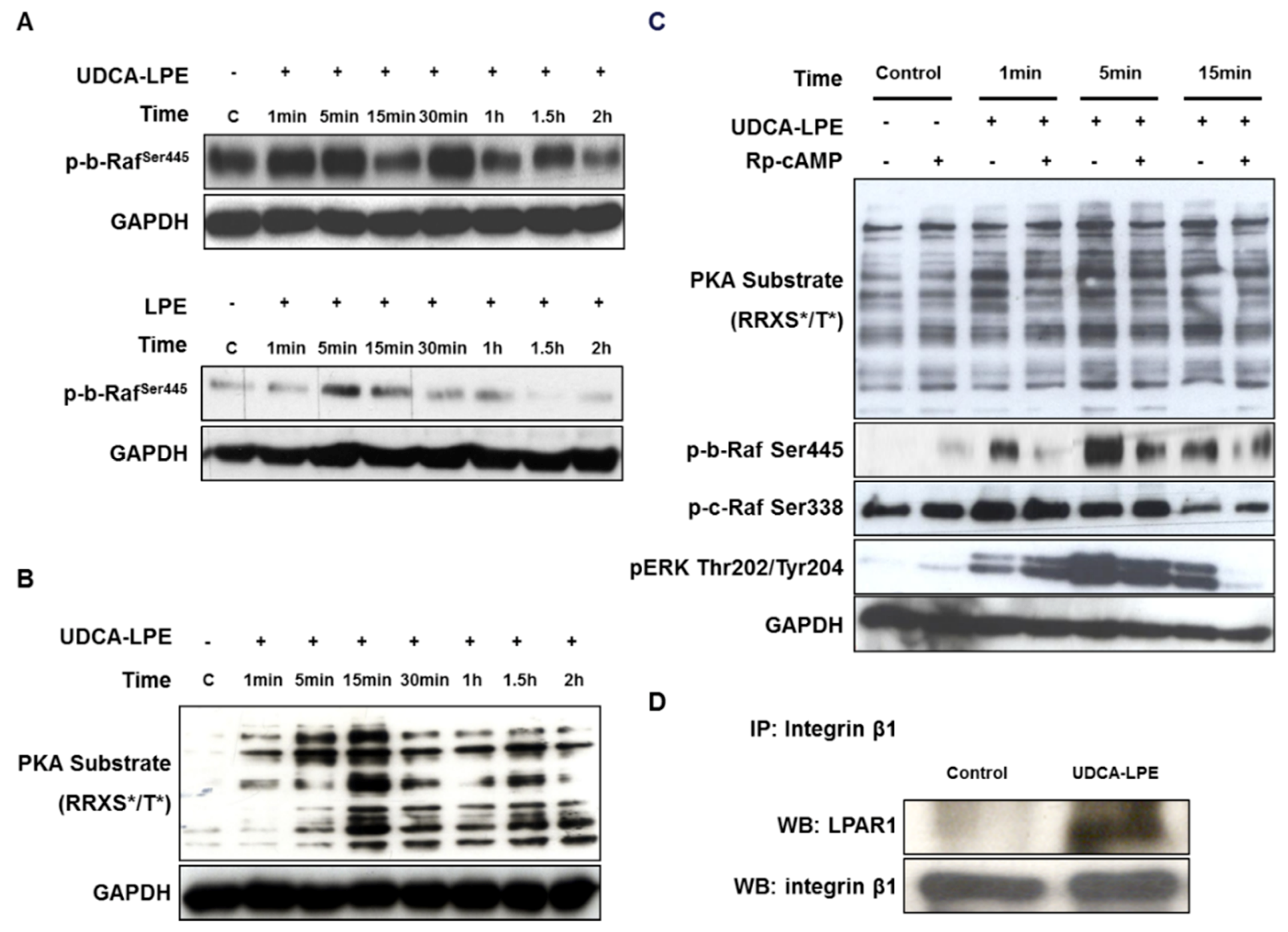
2.5. UDCA-LPE Serves as a Bivalent Ligand Bridging Between Integrin β1 and LPAR1
2.6. LPE-Moiety is Necessary for UDCA-LPE-Induced Translocation of Integrin β1 and Suppressed FAK and SRC Phosphorylation
2.7. UDCA-LPE Mediates the Compartmentalization of Integrins into Lipid Rafts
2.8. Integrin-Bound UDCA-LPE Translocated into Lipid Rafts, Which Co-Fractionated with LPE but Not UDCA
3. Discussion
4. Materials and Methods
4.1. Reagents and Cell Culture
4.2. Western Blotting
4.3. Immunofluorescence
4.4. Lipid Fractionation
4.5. Quantification of UDCA-LPE, UDCA and LPE
4.6. Immunoprecipitation
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ECM | extracellular matrix |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FAK | focal adhesion kinase |
| HHStec | primary human hepatic stellate cells |
| LPAR1 | lysophosphatidic acid receptor 1 |
| LPE | lysophosphatidylethanolamine |
| RGD | L-arginine, glycine and L-aspartic acid |
| TUDCA | tauro-ursodeoxycholic acid |
| UDCA | ursodeoxycholic acid |
| UDCA-LPE | ursodeoxycholyl lysophosphatidylethanolamide |
References
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Patsenker, E.; Popov, Y.; Wiesner, M.; Goodman, S.L.; Schuppan, D. Pharmacological inhibition of the vitronectin receptor abrogates PDGF-BB-induced hepatic stellate cell migration and activation in vitro. J. Hepatol. 2007, 46, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Chamulitrat, W.; Burhenne, J.; Rehlen, T.; Pathil, A.; Stremmel, W. Bile salt-phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamide as a hepatoprotective agent. Hepatology 2009, 50, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathil, A.; Mueller, J.; Ludwig, J.M.; Wang, J.; Warth, A.; Chamulitrat, W.; Stremmel, W. Ursodeoxycholyl lysophosphatidylethanolamide attenuates hepatofibrogenesis by impairment of TGF-β1/SMAD2/3 signalling. Br. J. Pharmacol. 2014, 171, 5113–5126. [Google Scholar] [CrossRef] [PubMed]
- Roma, M.G.; Toledo, F.D.; Boaglio, A.C.; Basiglio, C.L.; Crocenzi, F.A.; Sanchez Pozzi, E.J. Ursodeoxycholic acid in cholestasis: Linking action mechanisms to therapeutic applications. Clin. Sci. 2011, 121, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Poupon, R.E.; Lindor, K.D.; Pares, A.; Chazouilleres, O.; Poupon, R.; Heathcote, E.J. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J. Hepatol. 2003, 39, 12–16. [Google Scholar] [CrossRef]
- Pathil, A.; Warth, A.; Chamulitrat, W.; Stremmel, W. The synthetic bile acid-phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamide suppresses TNFα-induced liver injury. J. Hepatol. 2011, 54, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Esteves, M.; Ferreira, M.J.; Kozica, A.; Fernandes, A.C.; Goncalves da Silva, A.; Saramago, B. Interaction of cytotoxic and cytoprotective bile acids with model membranes: Influence of the membrane composition. Langmuir 2015, 31, 8901–8910. [Google Scholar] [CrossRef] [PubMed]
- Fahey, D.A.; Carey, M.C.; Donovan, J.M. Bile acid/phosphatidylcholine interactions in mixed monomolecular layers: Differences in condensation effects but not interfacial orientation between hydrophobic and hydrophilic bile acid species. Biochemistry 1995, 34, 10886–10897. [Google Scholar] [CrossRef] [PubMed]
- Escriba, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Torok, Z.; Horvath, I.; Harwood, J.L.; Vigh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-SRC signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.J.; Takashima, M.; Rippe, R.A. Molecular mechanisms of hepatic fibrogenesis. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. 1), S79–S84. [Google Scholar] [CrossRef]
- Wu, H.J.; Zhang, Z.Q.; Yu, B.; Liu, S.; Qin, K.R.; Zhu, L. Pressure activates Src-dependent FAK-AKT and ERK1/2 signaling pathways in rat hepatic stellate cells. Cell. Physiol. Biochem. 2010, 26, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A.; Golbik, R.; Mann, K.; Kuhn, K. The alpha 1 beta 1 integrin recognition site of the basement membrane collagen molecule [alpha 1(iv)]2 alpha 2(iv). EMBO J. 1993, 12, 4795–4802. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Lee, K.P.; Kang, S.; Chung, H.Y.; Bae, Y.S.; Okajima, F.; Im, D.S. Lysophosphatidylethanolamine utilizes LPA and CD97 in MDA-MB-231 breast cancer cells. Cell. Signal. 2013, 25, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Jang, I.S.; Rhim, J.H.; Park, S.C.; Yeo, E.J. Downstream molecular events in the altered profiles of lysophosphatidic acid-induced camp in senescent human diploid fibroblasts. Exp. Mol. Med. 2006, 38, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Spohr, T.C.; Dezonne, R.S.; Rehen, S.K.; Gomes, F.C. LPA-primed astrocytes induce axonal outgrowth of cortical progenitors by activating PKA signaling pathways and modulating extracellular matrix proteins. Front. Cell. Neurosci. 2014, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Jalink, K.; Hengeveld, T.; Mulder, S.; Postma, F.R.; Simon, M.F.; Chap, H.; van der Marel, G.A.; van Boom, J.H.; van Blitterswijk, W.J.; Moolenaar, W.H. Lysophosphatidic acid-induced Ca2+ mobilization in human A431 cells: Structure-activity analysis. Biochem. J. 1995, 307, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Bandoh, K.; Aoki, J.; Taira, A.; Tsujimoto, M.; Arai, H.; Inoue, K. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-activity relationship of cloned LPA receptors. FEBS Lett. 2000, 478, 159–165. [Google Scholar] [CrossRef]
- Harikumar, K.G.; Akgun, E.; Portoghese, P.S.; Miller, L.J. Modulation of cell surface expression of nonactivated cholecystokinin receptors using bivalent ligand-induced internalization. J. Med. Chem. 2010, 53, 2836–2842. [Google Scholar] [CrossRef] [PubMed]
- Schliess, F.; Kurz, A.K.; vom Dahl, S.; Haussinger, D. Mitogen-activated protein kinases mediate the stimulation of bile acid secretion by tauroursodeoxycholate in rat liver. Gastroenterology 1997, 113, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Haussinger, D.; Kurz, A.K.; Wettstein, M.; Graf, D.; Vom Dahl, S.; Schliess, F. Involvement of integrins and src in tauroursodeoxycholate-induced and swelling-induced choleresis. Gastroenterology 2003, 124, 1476–1487. [Google Scholar] [CrossRef]
- Gohlke, H.; Schmitz, B.; Sommerfeld, A.; Reinehr, R.; Haussinger, D. Alpha5 beta1-integrins are sensors for tauroursodeoxycholic acid in hepatocytes. Hepatology 2013, 57, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Mello-Vieira, J.; Sousa, T.; Coutinho, A.; Fedorov, A.; Lucas, S.D.; Moreira, R.; Castro, R.E.; Rodrigues, C.M.; Prieto, M.; Fernandes, F. Cytotoxic bile acids, but not cytoprotective species, inhibit the ordering effect of cholesterol in model membranes at physiologically active concentrations. Biochim. Biophys. Acta 2013, 1828, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Ben Mouaz, A.; Lindheimer, M.; Montet, J.C.; Zajac, J.; Lagerge, S. A study of the adsorption of bile salts onto model lecithin membranes. Colloids Surf. B Biointerfaces 2001, 20, 119–127. [Google Scholar] [CrossRef]
- Hiller, C.; Kuhhorn, J.; Gmeiner, P. Class a G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J. Med. Chem. 2013, 56, 6542–6559. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, F., Jr.; Bernier, S.G.; Vazquez-Tello, A.; Brault, S.; Beauchamp, M.H.; Quiniou, C.; Marrache, A.M.; Checchin, D.; Sennlaub, F.; Hou, X.; et al. Modulation of pro-inflammatory gene expression by nuclear lysophosphatidic acid receptor type-1. J. Biol. Chem. 2003, 278, 38875–38883. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, D.; Su, Y.; Berdyshev, E.; Chun, J.; Natarajan, V.; Zhao, Y. Lysophosphatidic acid receptor 1 modulates lipopolysaccharide-induced inflammation in alveolar epithelial cells and murine lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L547–L556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peres, C.; Yart, A.; Perret, B.; Salles, J.P.; Raynal, P. Modulation of phosphoinositide 3-kinase activation by cholesterol level suggests a novel positive role for lipid rafts in lysophosphatidic acid signalling. FEBS Lett. 2003, 534, 164–168. [Google Scholar] [CrossRef] [Green Version]
- Heuman, D.M.; Bajaj, R. Ursodeoxycholate conjugates protect against disruption of cholesterol-rich membranes by bile salts. Gastroenterology 1994, 106, 1333–1341. [Google Scholar] [CrossRef]
- Shi, F.; Sottile, J. Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 2008, 121, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Heino, J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 2011, 27, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Hedin, U.L.; Daum, G.; Clowes, A.W. Disruption of integrin alpha 5 beta 1 signaling does not impair PDGF-BB-mediated stimulation of the extracellular signal-regulated kinase pathway in smooth muscle cells. J. Cell. Physiol. 1997, 172, 109–116. [Google Scholar] [CrossRef]
- Horton, M.A.; Taylor, M.L.; Arnett, T.R.; Helfrich, M.H. Arg-Gly-Asp (RGD) peptides and the anti-vitronectin receptor antibody 23C6 inhibit dentine resorption and cell spreading by osteoclasts. Exp. Cell Res. 1991, 195, 368–375. [Google Scholar] [CrossRef]
- Ylanne, J. Rgd peptides may only temporarily inhibit cell adhesion to fibronectin. FEBS Lett. 1990, 267, 43–45. [Google Scholar] [CrossRef]
- Katow, H.; Yazawa, S.; Sofuku, S. A fibronectin-related synthetic peptide, Pro-Ala-Ser-Ser, inhibits fibronectin binding to the cell surface, fibronectin-promoted cell migration in vitro, and cell migration in vivo. Exp. Cell Res. 1990, 190, 17–24. [Google Scholar] [CrossRef]
- Staubli, U.; Chun, D.; Lynch, G. Time-dependent reversal of long-term potentiation by an integrin antagonist. J. Neurosci. 1998, 18, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Focal adhesion kinase: A key mediator of transforming growth factor beta signaling in fibroblasts. Adv. Wound Care 2013, 2, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Gunther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.P.; Plow, E.F.; Frelinger, A.L.; O’Toole, T.E.; Loftus, J.C.; Ginsberg, M.H. Ligands “activate” integrin alpha IIb beta 3 (platelet GPIIb-IIIa). Cell 1991, 65, 409–416. [Google Scholar] [CrossRef]
- Mayo, K.H.; Fan, F.; Beavers, M.P.; Eckardt, A.; Keane, P.; Hoekstra, W.J.; Andrade-Gordon, P. Rgd induces conformational transition in purified platelet integrin GPIIb/IIIa-SDS system yielding multiple binding states for fibrinogen gamma-chain C-terminal peptide. FEBS Lett. 1996, 378, 79–82. [Google Scholar] [CrossRef]
- Jiao, L.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Chamulitrat, W. Sensitization to autoimmune hepatitis in group VIA calcium-independent phospholipase A2-null mice led to duodenal villous atrophy with apoptosis, goblet cell hyperplasia and leaked bile acids. Biochim. Biophys. Acta 2015, 1852, 1646–1657. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, J.; Gan-Schreier, H.; Goeppert, B.; Chamulitrat, W.; Stremmel, W.; Pathil, A. Bivalent Ligand UDCA-LPE Inhibits Pro-Fibrogenic Integrin Signalling by Inducing Lipid Raft-Mediated Internalization. Int. J. Mol. Sci. 2018, 19, 3254. https://doi.org/10.3390/ijms19103254
Su J, Gan-Schreier H, Goeppert B, Chamulitrat W, Stremmel W, Pathil A. Bivalent Ligand UDCA-LPE Inhibits Pro-Fibrogenic Integrin Signalling by Inducing Lipid Raft-Mediated Internalization. International Journal of Molecular Sciences. 2018; 19(10):3254. https://doi.org/10.3390/ijms19103254
Chicago/Turabian StyleSu, Jie, Hongying Gan-Schreier, Benjamin Goeppert, Walee Chamulitrat, Wolfgang Stremmel, and Anita Pathil. 2018. "Bivalent Ligand UDCA-LPE Inhibits Pro-Fibrogenic Integrin Signalling by Inducing Lipid Raft-Mediated Internalization" International Journal of Molecular Sciences 19, no. 10: 3254. https://doi.org/10.3390/ijms19103254
APA StyleSu, J., Gan-Schreier, H., Goeppert, B., Chamulitrat, W., Stremmel, W., & Pathil, A. (2018). Bivalent Ligand UDCA-LPE Inhibits Pro-Fibrogenic Integrin Signalling by Inducing Lipid Raft-Mediated Internalization. International Journal of Molecular Sciences, 19(10), 3254. https://doi.org/10.3390/ijms19103254