Detours to Replication: Functions of Specialized DNA Polymerases during Oncogene-induced Replication Stress


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Endogenous Genome DiToRS
2.1. AT-Rich Repeats
2.2. GC-Rich Repeats
2.3. Triplex DNA (H-DNA)
2.4. Inverted Repeats and Quasipalindromes
3. Specialized DNA Polymerases and the Maintenance of DiToRS Stability
3.1. Specialized Polymerases and Common Fragile Site Replication
3.2. Specialized Polymerase Synthesis of G4 Motifs
4. Specialized DNA Polymerases and the Replication Stress Response
5. Oncogenic Replication Stress Mechanisms
5.1. Balancing Cell Proliferation, Apoptosis, and Cell Death
5.2. Regulation of DNA Replication and S Phase
5.3. Alterations in Metabolism
6. Tolerance of Oncogenic Replication Stress via Specialized DNA Polymerases
7. Perspective
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CFS | Common Fragile Site |
| DiToRS | Difficult-to-Replicate Sequences |
| Non-B DNA | DNA secondary structures other than B form |
References
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microb. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Koundrioukoff, S.; Wilhelm, T.; Letessier, A.; Brison, O.; Debatisse, M. Updating the mechanisms of common fragile site instability: How to reconcile the different views? Cell. Mol. Life Sci. 2014, 71, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freudenreich, C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. A structure for deoxyribose nucleic acid. 1953. Nature 2003, 421, 397–398. [Google Scholar] [PubMed]
- Wang, A.H.; Quigley, G.J.; Kolpak, F.J.; Crawford, J.L.; van Boom, J.H.; van der Marel, G.; Rich, A. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature 1979, 282, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M.; Lyamichev, V.I.; Drushlyak, K.N.; Dobrynin, V.N.; Filippov, S.A.; Frank-Kamenetskii, M.D. DNA H form requires a homopurine-homopyrimidine mirror repeat. Nature 1987, 330, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Schroth, G.P.; Ho, P.S. Occurrence of potential cruciform and H-DNA forming sequences in genomic DNA. Nucleic Acids Res. 1995, 23, 1977–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, G.N.; Lee, M.P.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.S.; Wu, H.M.; Crothers, D.M. DNA bending at adenine. thymine tracts. Nature 1986, 320, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L. Structure, location and interactions of G-quadruplexes. FEBS J. 2010, 277, 3452–3458. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, H.; Ohye, T.; Kogo, H.; Kato, T.; Bolor, H.; Taniguchi, M.; Shaikh, T.H.; Emanuel, B.S.; Kurahashi, H. Chromosomal instability mediated by non-B DNA: Cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res. 2009, 19, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Bissler, J.J. DNA inverted repeats and human disease. Front. Biosci. 1998, 3, d408–d418. [Google Scholar] [CrossRef] [PubMed]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.; Wang, Y.H. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genom. 2015, 16, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Pierce, L.J.; Spangrude, G.J. Distinct roles of IL-7 and stem cell factor in the OP9-DL1 T-cell differentiation culture system. Exp. Hematol. 2006, 34, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Carbajal, S.; Vijg, J.; DiGiovanni, J.; Vasquez, K.M. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008, 100, 1815–1817. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.J.; Mirkin, S.M. Cis- and Trans-Modifiers of Repeat Expansions: Blending Model Systems with Human Genetics. Trends Genet. 2018, 34, 448–465. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Carvalho, C.M.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Colnaghi, R.; Carpenter, G.; Volker, M.; O’Driscoll, M. The consequences of structural genomic alterations in humans: Genomic disorders, genomic instability and cancer. Semin. Cell Dev. Biol. 2011, 22, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Tainer, J.A.; Vasquez, K.M.; Cooper, D.N. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 2016, 44, 5673–5688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrow, A.A.; Williams, L.E.; Pierce, L.C.; Wang, Y.H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genom. 2009, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, G.; Del Mundo, I.M.; McKinney, J.A.; Lu, X.; Bacolla, A.; Boulware, S.B.; Zhang, C.; Zhang, H.; Ren, P.; et al. Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes. Cell Rep. 2018, 22, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S. Repetitive DNA in eukaryotic genomes. Chromosome Res. 2015, 23, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Gemayel, R.; Vinces, M.D.; Legendre, M.; Verstrepen, K.J. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 2010, 44, 445–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Zlotorynski, E.; Rahat, A.; Skaug, J.; Ben-Porat, N.; Ozeri, E.; Hershberg, R.; Levi, A.; Scherer, S.W.; Margalit, H.; Kerem, B. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol. 2003, 23, 7143–7151. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Stout-Weider, K.; Hermann, K.; Schrock, E.; Heiden, T. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res. 2006, 16, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoder, C.; Liehr, T.; Velleuer, E.; Wilhelm, K.; Blaurock, N.; Weise, A.; Mrasek, K. New aspects on chromosomal instability: Chromosomal break-points in Fanconi anemia patients co-localize on the molecular level with fragile sites. Int. J. Oncol. 2010, 36, 307–312. [Google Scholar] [PubMed]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Narva, E.; Ng, S.; Sourour, M.; Hamalainen, R.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011, 471, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Dall, K.L.; Scarpini, C.G.; Roberts, I.; Winder, D.M.; Stanley, M.A.; Muralidhar, B.; Herdman, M.T.; Pett, M.R.; Coleman, N. Characterization of naturally occurring HPV16 integration sites isolated from cervical keratinocytes under noncompetitive conditions. Cancer Res. 2008, 68, 8249–8259. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Schwartz, M.; Schmidt, M.; Garrigue, A.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M.; Ben-Porat, N.; Von Kalle, C.; Fischer, A.; Kerem, B. Fragile sites are preferential targets for integrations of MLV vectors in gene therapy. Gene Ther. 2006, 13, 1057–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, L.W.; Burrow, A.A.; Wang, Y.H. DNA instability at chromosomal fragile sites in cancer. Curr. Genom. 2010, 11, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Lukusa, T.; Fryns, J.P. Human chromosome fragility. Biochim. Biophys. Acta 2008, 1779, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Lebofsky, R.; Rahat, A.; Bester, A.C.; Bensimon, A.; Kerem, B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 2011, 43, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Palakodeti, A.; Lucas, I.; Jiang, Y.; Young, D.J.; Fernald, A.A.; Karrison, T.; Le Beau, M.M. Impaired replication dynamics at the FRA3B common fragile site. Hum. Mol. Genet. 2010, 19, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Fungtammasan, A.; Walsh, E.; Chiaromonte, F.; Eckert, K.A.; Makova, K.D. A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res. 2012, 22, 993–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishmar, D.; Rahat, A.; Scherer, S.W.; Nyakatura, G.; Hinzmann, B.; Kohwi, Y.; Mandel-Gutfroind, Y.; Lee, J.R.; Drescher, B.; Sas, D.E.; et al. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc. Natl. Acad. Sci. USA 1998, 95, 8141–8146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrow, A.A.; Marullo, A.; Holder, L.R.; Wang, Y.H. Secondary structure formation and DNA instability at fragile site FRA16B. Nucleic Acids Res. 2010, 38, 2865–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.N.; Opresko, P.L.; Meng, X.; Lee, M.Y.; Eckert, K.A. DNA structure and the Werner protein modulate human DNA polymerase Delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 2010, 38, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.; Wang, X.; Lee, M.Y.; Eckert, K.A. Mechanism of replicative DNA polymerase Delta pausing and a potential role for DNA polymerase Kappa in common fragile site replication. J. Mol. Biol. 2013, 425, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Hile, S.E.; Eckert, K.A. DNA polymerase kappa produces interrupted mutations and displays polar pausing within mononucleotide microsatellite sequences. Nucleic Acids Res. 2008, 36, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.P.; Hile, S.E.; Lee, M.Y.; Eckert, K.A. DNA polymerases Eta and Kappa exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair (Amst) 2017, 57, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; House, N.C.; Freudenreich, C.H. Repeat instability during DNA repair: Insights from model systems. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 142–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usdin, K.; Woodford, K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995, 23, 4202–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitzmann, M.N.; Woodford, K.J.; Usdin, K. The development and use of a DNA polymerase arrest assay for the evaluation of parameters affecting intrastrand tetraplex formation. J. Biol. Chem. 1996, 271, 20958–20964. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.; Krasilnikova, M.M.; Samadashwily, G.M.; Lahue, R.; Mirkin, S.M. Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 2003, 23, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Surka, C.F.; Shishkin, A.A.; Krasilnikova, M.M.; Mirkin, S.M. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009, 16, 226–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lormand, J.D.; Buncher, N.; Murphy, C.T.; Kaur, P.; Lee, M.Y.; Burgers, P.; Wang, H.; Kunkel, T.A.; Opresko, P.L. DNA polymerase delta stalls on telomeric lagging strand templates independently from G-quadruplex formation. Nucleic Acids Res. 2013, 41, 10323–10333. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Michor, F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat. Struct. Mol. Biol. 2011, 18, 950–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Vasquez, K.M. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13448–13453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hile, S.E.; Eckert, K.A. Positive correlation between DNA polymerase alpha-primase pausing and mutagenesis within polypyrimidine/polypurine microsatellite sequences. J. Mol. Biol. 2004, 335, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikova, M.M.; Mirkin, S.M. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol. Cell. Biol. 2004, 24, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Chandok, G.S.; Patel, M.P.; Mirkin, S.M.; Krasilnikova, M.M. Effects of Friedreich’s ataxia GAA repeats on DNA replication in mammalian cells. Nucleic Acids Res. 2012, 40, 3964–3974. [Google Scholar] [CrossRef] [PubMed]
- Follonier, C.; Oehler, J.; Herrador, R.; Lopes, M. Friedreich’s ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat. Struct. Mol. Biol. 2013, 20, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Casper, A.M.; Greenwell, P.W.; Tang, W.; Petes, T.D. Chromosome aberrations resulting from double-strand DNA breaks at a naturally occurring yeast fragile site composed of inverted ty elements are independent of Mre11p and Sae2p. Genetics 2009, 183, 423–439, 1SI–26SI. [Google Scholar] [CrossRef] [PubMed]
- Seier, T.; Padgett, D.R.; Zilberberg, G.; Sutera, V.A., Jr.; Toha, N.; Lovett, S.T. Insights into mutagenesis using Escherichia coli chromosomal lacZ strains that enable detection of a wide spectrum of mutational events. Genetics 2011, 188, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Narayanan, V.; Lobachev, K.S.; Mirkin, S.M. Replication stalling at unstable inverted repeats: Interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 9936–9941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Wang, G.; Bacolla, A.; Zhao, J.; Spitser, S.; Vasquez, K.M. Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep. 2015, 10, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microb. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed]
- Hile, S.E.; Wang, X.; Lee, M.Y.; Eckert, K.A. Beyond translesion synthesis: Polymerase κ fidelity as a potential determinant of microsatellite stability. Nucleic Acids Res. 2012, 40, 1636–1647. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.; Eckert, K. Maintenance of Genome Integrity: How Mammalian Cells Orchestrate Genome Duplication by Coordinating Replicative and Specialized DNA Polymerases. Genes (Basel) 2017, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Bournique, E.; Dall’Osto, M.; Hoffmann, J.S.; Bergoglio, V. Role of specialized DNA polymerases in the limitation of replicative stress and DNA damage transmission. Mutat. Res. 2018, 808, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.S.; Grgurevic, S.; Cazaux, C.; Hoffmann, J.S. The human specialized DNA polymerases and non-B DNA: Vital relationships to preserve genome integrity. J. Mol. Biol. 2013, 425, 4767–4781. [Google Scholar] [CrossRef] [PubMed]
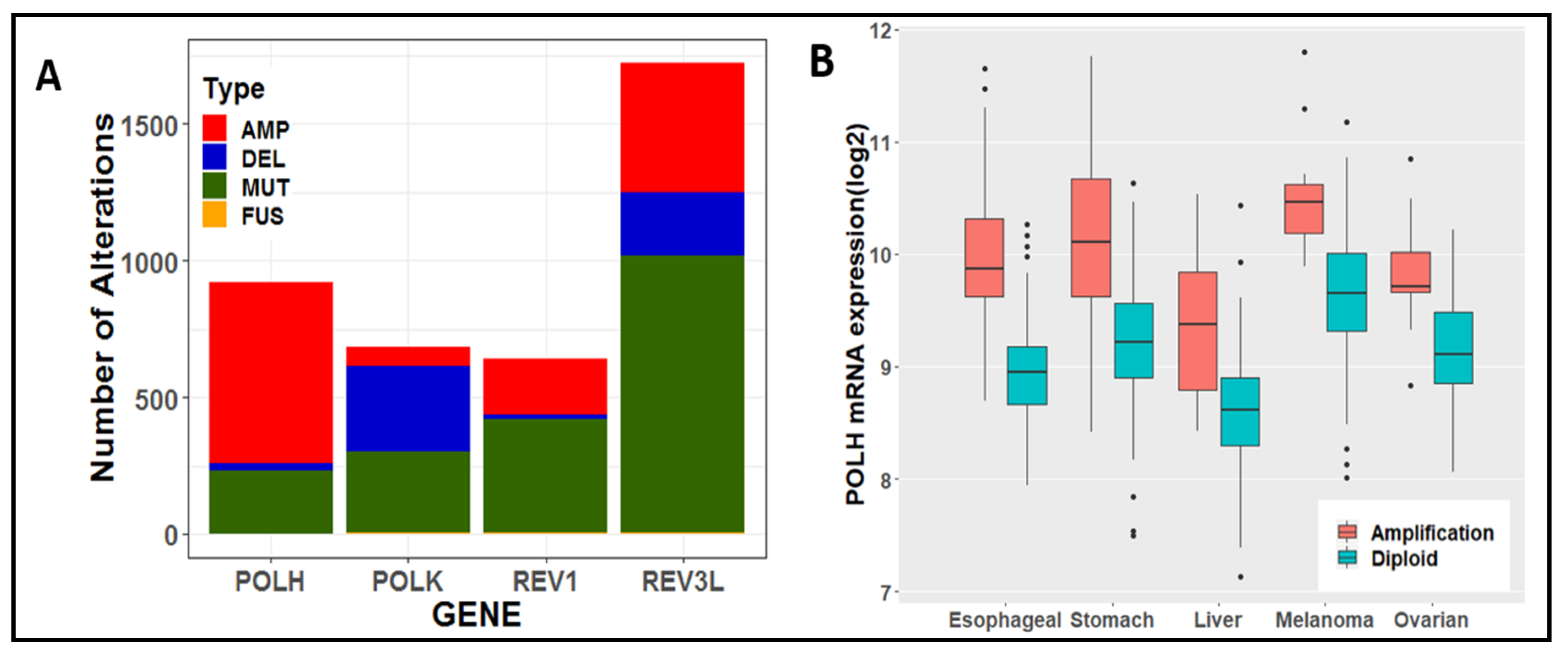
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013, 6, l1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceppi, P.; Novello, S.; Cambieri, A.; Longo, M.; Monica, V.; Lo Iacono, M.; Giaj-Levra, M.; Saviozzi, S.; Volante, M.; Papotti, M.; et al. Polymerase eta mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res. 2009, 15, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, Y.W.; Liu, X.; Chu, P.; Loria, S.; Wang, Y.; Yen, Y.; Chou, K.M. Expression of DNA translesion synthesis polymerase eta in head and neck squamous cell cancer predicts resistance to gemcitabine and cisplatin-based chemotherapy. PLoS ONE 2013, 8, e83978. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.C.; Cordonnier, A.; Kleijer, W.J.; Jaspers, N.G.; Fawcett, H.; Raams, A.; Garritsen, V.H.; Stary, A.; Avril, M.F.; Boudsocq, F.; et al. Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients. Proc. Natl. Acad. Sci. USA 2002, 99, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Iwai, S.; Hanaoka, F. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. EMBO J. 2000, 19, 3100–3109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biertümpfel, C.; Zhao, Y.; Kondo, Y.; Ramón-Maiques, S.; Gregory, M.; Lee, J.Y.; Masutani, C.; Lehmann, A.R.; Hanaoka, F.; Yang, W. Structure and mechanism of human DNA polymerase eta. Nature 2010, 465, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Stary, A.; Kannouche, P.; Lehmann, A.R.; Sarasin, A. Role of DNA polymerase eta in the UV mutation spectrum in human cells. J. Biol. Chem. 2003, 278, 18767–18775. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Maher, V.M.; McCormick, J.J. Xeroderma pigmentosum variant cells are less likely than normal cells to incorporate dAMP opposite photoproducts during replication of UV-irradiated plasmids. Proc. Natl. Acad. Sci. USA 1991, 88, 7810–7814. [Google Scholar] [CrossRef] [PubMed]
- Peña-Diaz, J.; Bregenhorn, S.; Ghodgaonkar, M.; Follonier, C.; Artola-Borán, M.; Castor, D.; Lopes, M.; Sartori, A.A.; Jiricny, J. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol. Cell 2012, 47, 669–680. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, M.J.; Mcllwraith, M.J.; Vaisman, A.; Liu, Y.; Fanning, E.; Woodgate, R.; West, S.C. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol. Cell 2005, 20, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Winter, D.B.; Kasmer, C.; Kraemer, K.H.; Lehmann, A.R.; Gearhart, P.J. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2001, 2, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Goncearenco, A.; Lada, A.G.; De, S.; Yurchenko, V.; Nudelman, G.; Panchenko, A.R.; Cooper, D.N.; Pavlov, Y.I. DNA polymerase η mutational signatures are found in a variety of different types of cancer. Cell Cycle 2018, 17, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Albertella, M.R.; Lau, A.; O’Connor, M.J. The overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst) 2005, 4, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Fang, Y.; Xu, Y.; Zhang, K.; Hu, X. Down-regulation of DNA polymerases κ, η, ι, and ζ in human lung, stomach, and colorectal cancers. Cancer Lett. 2005, 217, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Pillaire, M.J.; Selves, J.; Gordien, K.; Gourraud, P.A.; Gouraud, P.A.; Gentil, C.; Danjoux, M.; Do, C.; Negre, V.; Bieth, A.; et al. A ‘DNA replication’ signature of progression and negative outcome in colorectal cancer. Oncogene 2010, 29, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.; Townson, S.A.; Uljon, S.N.; Johnson, R.E.; Brahma, A.; Nair, D.T.; Prakash, S.; Prakash, L.; Aggarwal, A.K. Human DNA polymerase Kappa encircles DNA: Implications for mismatch extension and lesion bypass. Mol. Cell 2007, 25, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Ogi, T.; Lehmann, A.R. The Y-family DNA polymerase κ (pol κ) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 2006, 8, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lv, L.; Chen, Q.; Yuan, F.; Zhang, T.; Yang, Y.; Zhang, H.; Wang, Y.; Jia, Y.; Qian, L.; et al. Mouse DNA polymerase Kappa has a functional role in the repair of DNA strand breaks. DNA Repair (Amst) 2013, 12, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Avkin, S.; Goldsmith, M.; Velasco-Miguel, S.; Geacintov, N.; Friedberg, E.C.; Livneh, Z. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: The role of DNA polymerase kappa. J. Biol. Chem. 2004, 279, 53298–53305. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Slater, D.M.; Ohmori, H.; Vaziri, C. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J. Biol. Chem. 2005, 280, 22343–22355. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Bebenek, K.; Matsuda, T.; Feaver, W.J.; Gerlach, V.L.; Friedberg, E.C.; Ohmori, H.; Kunkel, T.A. Fidelity and processivity of DNA synthesis by DNA polymerase κ, the product of the human DINB1 gene. J. Biol. Chem. 2000, 275, 39678–39684. [Google Scholar] [CrossRef] [PubMed]
- Bavoux, C.; Leopoldino, A.M.; Bergoglio, V.J.O.W.; Ogi, T.; Bieth, A.; Judde, J.G.; Pena, S.D.; Poupon, M.F.; Helleday, T.; et al. Up-regulation of the error-prone DNA polymerase κ promotes pleiotropic genetic alterations and tumorigenesis. Cancer Res. 2005, 65, 325–330. [Google Scholar] [PubMed]
- Doles, J.; Oliver, T.G.; Cameron, E.R.; Hsu, G.; Jacks, T.; Walker, G.C.; Hemann, M.T. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc. Natl. Acad. Sci. USA 2010, 107, 20786–20791. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Deoxycytidyl transferase activity of yeast REV1 protein. Nature 1996, 382, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, X.; Rechkoblit, O.; Geacintov, N.E.; Taylor, J.S.; Wang, Z. Response of human REV1 to different DNA damage: Preferential dCMP insertion opposite the lesion. Nucleic Acids Res. 2002, 30, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.T.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science 2005, 309, 2219–2222. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.L.; Simpson, L.J.; Sale, J.E. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005, 33, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Fischhaber, P.L.; Luk-Paszyc, M.J.; Masuda, Y.; Zhou, J.; Kamiya, K.; Kisker, C.; Friedberg, E.C. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003, 22, 6621–6630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, E.; Murakumo, Y.; Kanjo, N.; Akagi, J.; Masutani, C.; Hanaoka, F.; Ohmori, H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells 2004, 9, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Gregory, M.T.; Yang, W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc. Natl. Acad. Sci. USA 2014, 111, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedberg, E.C.; Fischhaber, P.L.; Kisker, C. Error-prone DNA polymerases: Novel structures and the benefits of infidelity. Cell 2001, 107, 9–12. [Google Scholar] [CrossRef]
- Diaz, M.; Watson, N.B.; Turkington, G.; Verkoczy, L.K.; Klinman, N.R.; McGregor, W.G. Decreased frequency and highly aberrant spectrum of ultraviolet-induced mutations in the hprt gene of mouse fibroblasts expressing antisense RNA to DNA polymerase zeta. Mol. Cancer Res. 2003, 1, 836–847. [Google Scholar] [PubMed]
- Despras, E.; Sittewelle, M.; Pouvelle, C.; Delrieu, N.; Cordonnier, A.M.; Kannouche, P.L. Rad18-dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat. Commun. 2016, 7, 13326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, L.; Sidorova, J.M.; Puget, N.; Boudsocq, F.; Biard, D.S.; Monnat, R.J., Jr.; Cazaux, C.; Hoffmann, J.S. Human DNA polymerase eta is required for common fragile site stability during unperturbed DNA replication. Mol. Cell. Biol. 2009, 29, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Niraj, J.; Pauty, J.; Maity, R.; Zhao, W.; Coulombe, Y.; Sung, P.; Masson, J.Y. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase eta in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014, 6, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Baptiste, B.A.; Eckert, K.A. DNA polymerase kappa microsatellite synthesis: Two distinct mechanisms of slippage-mediated errors. Environ. Mol. Mutagen. 2012, 53, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, A.; Sridharan, S.; van Wietmarschen, N.; Maman, Y.; Callen, E.; Stanlie, A.; Wu, W.; Wu, X.; Day, A.; Wong, N.; et al. Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 2018, 174, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, S.F.; Bertolin, A.P.; Bergoglio, V.; Pillaire, M.J.; Gonzalez Besteiro, M.A.; Luzzani, C.; Miriuka, S.G.; Cazaux, C.; Hoffmann, J.S.; Gottifredi, V. Cyclin Kinase-independent role of p21(CDKN1A) in the promotion of nascent DNA elongation in unstressed cells. Elife 2016, 5, e18020. [Google Scholar] [CrossRef] [PubMed]
- Northam, M.R.; Robinson, H.A.; Kochenova, O.V.; Shcherbakova, P.V. Participation of DNA polymerase zeta in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics 2010, 184, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Northam, M.R.; Moore, E.A.; Mertz, T.M.; Binz, S.K.; Stith, C.M.; Stepchenkova, E.I.; Wendt, K.L.; Burgers, P.M.; Shcherbakova, P.V. DNA polymerases ζ and Rev1 mediate error-prone bypass of non-B DNA structures. Nucleic Acids Res. 2014, 42, 290–306. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.; Andersen, P.L.; Qin, Z.; Xiao, W. Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res. 2013, 41, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, C.M.; Arzouk, H.; Frey, A.; Maiter, A.; Sale, J.E. Contributions of the specialised DNA polymerases to replication of structured DNA. DNA Repair (Amst) 2015, 29, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Reams, C.; Simpson, L.J.; Sale, J.E. Epigenetic instability due to defective replication of structured DNA. Mol. Cell 2010, 40, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.G.; Spies, M. G-quadruplex recognition and remodeling by the FANCJ helicase. Nucleic Acids Res. 2016, 44, 8742–8753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkies, P.; Murat, P.; Phillips, L.G.; Patel, K.J.; Balasubramanian, S.; Sale, J.E. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2012, 40, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.; Maddukuri, L.; Ketkar, A.; Zafar, M.K.; Henninger, E.E.; Pursell, Z.F.; Eoff, R.L. Evidence for the kinetic partitioning of polymerase activity on G-quadruplex DNA. Biochemistry 2015, 54, 3218–3230. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.; Ketkar, A.; Zafar, M.K.; Maddukuri, L.; Choi, J.Y.; Eoff, R.L. Human Rev1 polymerase disrupts G-quadruplex DNA. Nucleic Acids Res. 2014, 42, 3272–3285. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.; Tillman, M.; Maddukuri, L.; Ketkar, A.; Zafar, M.K.; Eoff, R.L. Human Translesion Polymerase κ Exhibits Enhanced Activity and Reduced Fidelity Two Nucleotides from G-Quadruplex DNA. Biochemistry 2016, 55, 5218–5229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betous, R.; Rey, L.; Wang, G.; Pillaire, M.J.; Puget, N.; Selves, J.; Biard, D.S.; Shin-ya, K.; Vasquez, K.M.; Cazaux, C.; Hoffmann, J.S. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009, 48, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Exposito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-Gonzalez, J.; Zhang, S.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase η in the Alternative Lengthening of Telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef] [PubMed]
- Falabella, M.; Fernandez, R.J.; Johnson, B.; Kaufman, B.A. Potential roles for G-quadruplexes in mitochondria. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; Carneiro, J.; Goncalves, J.; Stewart, J.B.; Samuels, D.C.; Amorim, A.; Pereira, F. Mitochondrial DNA deletions are associated with non-B DNA conformations. Nucleic Acids Res. 2012, 40, 7606–7621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.W.; Pereira, F.; Barrett, S.P.; Kolesar, J.E.; Cao, K.; Damas, J.; Yatsunyk, L.A.; Johnson, F.B.; Kaufman, B.A. Association of G-quadruplex forming sequences with human mtDNA deletion breakpoints. BMC Genom. 2014, 15, 677. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; Zhou, J.; Kaplan, D.L.; Spelbrink, J.N.; Mergny, J.L.; Brosh, R.M., Jr. DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J. Biol. Chem. 2014, 289, 29975–29993. [Google Scholar] [CrossRef] [PubMed]
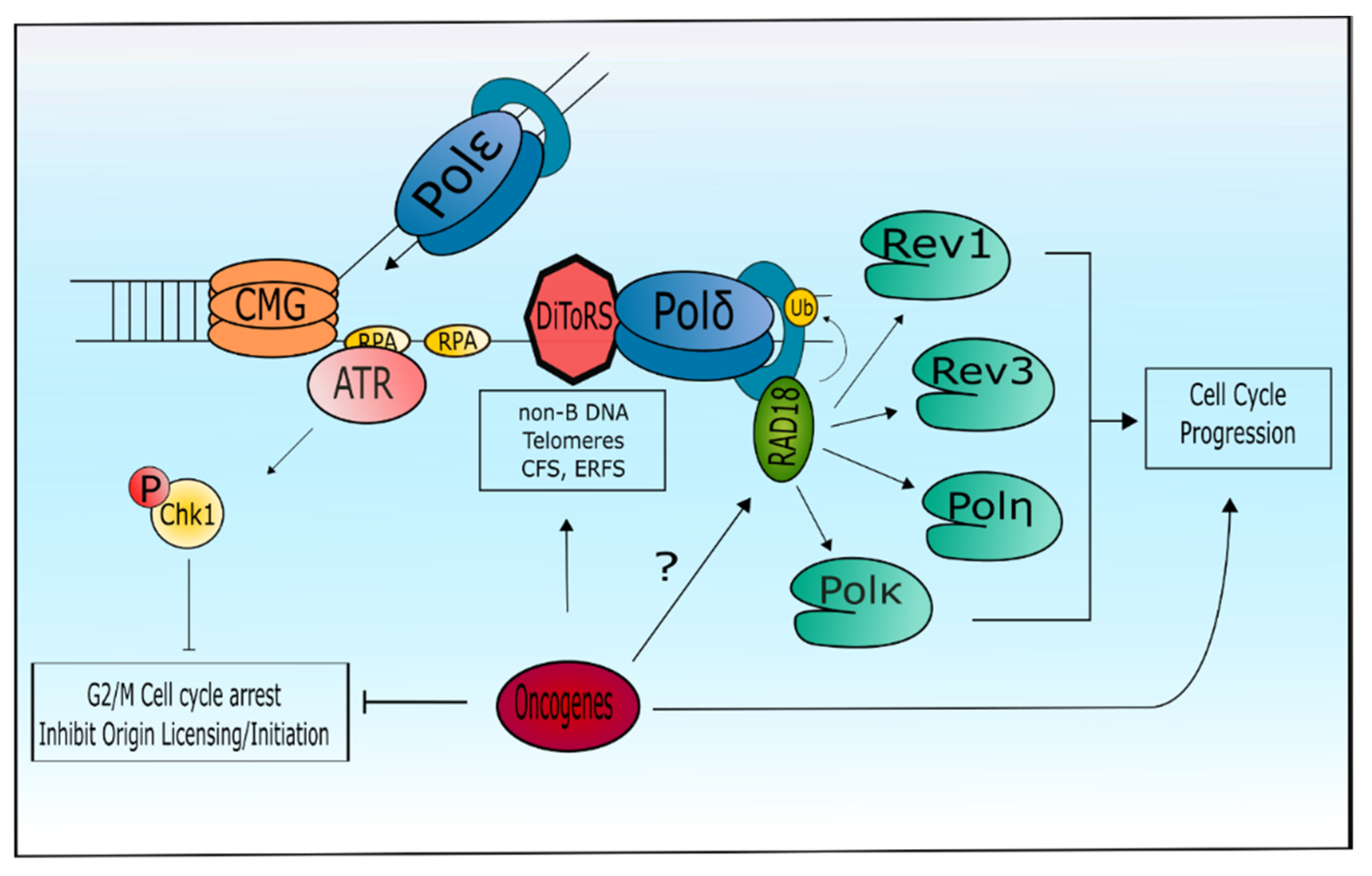
- Yan, S.; Michael, W.M. TopBP1 and DNA polymerase-α directly recruit the 9-1-1 complex to stalled DNA replication forks. J. Cell Biol. 2009, 184, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Bétous, R.; Pillaire, M.J.; Pierini, L.; van der Laan, S.; Recolin, B.; Ohl-Séguy, E.; Guo, C.; Niimi, N.; Grúz, P.; Nohmi, T.; et al. DNA polymerase κ-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 2013, 32, 2172–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeStephanis, D.; McLeod, M.; Yan, S. REV1 is important for the ATR-Chk1 DNA damage response pathway in Xenopus egg extracts. Biochem. Biophys. Res. Commun. 2015, 460, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4, a012955. [Google Scholar] [CrossRef] [PubMed]
- Futami, K.; Furuichi, Y. RECQL1 and WRN DNA repair helicases: Potential therapeutic targets and proliferative markers against cancers. Front. Genet. 2014, 5, 441. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomida, J.; Itaya, A.; Shigechi, T.; Unno, J.; Uchida, E.; Ikura, M.; Masuda, Y.; Matsuda, S.; Adachi, J.; Kobayashi, M.; et al. A novel interplay between the Fanconi anemia core complex and ATR-ATRIP kinase during DNA cross-link repair. Nucleic Acids Res. 2013, 41, 6930–6941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepal, M.; Che, R.; Zhang, J.; Ma, C.; Fei, P. Fanconi Anemia Signaling and Cancer. Trends Cancer 2017, 3, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Göhler, T.; Sabbioneda, S.; Green, C.M.; Lehmann, A.R. ATR-mediated phosphorylation of DNA polymerase η is needed for efficient recovery from UV damage. J. Cell Biol. 2011, 192, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Barkley, L.R.; Slater, D.M.; Tateishi, S.; Yamaizumi, M.; Ohmori, H.; Vaziri, C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell. Biol. 2006, 26, 3527–3540. [Google Scholar] [CrossRef] [PubMed]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef]
- Chen, Y.W.; Cleaver, J.E.; Hatahet, Z.; Honkanen, R.E.; Chang, J.Y.; Yen, Y.; Chou, K.M. Human DNA polymerase eta activity and translocation is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 16578–16583. [Google Scholar] [CrossRef] [PubMed]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell. Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Miron, K.; Golan-Lev, T.; Dvir, R.; Ben-David, E.; Kerem, B. Oncogenes create a unique landscape of fragile sites. Nat. Commun. 2015, 6, 7094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Shortt, J.; Johnstone, R.W. Oncogenes in cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a009829. [Google Scholar] [CrossRef] [PubMed]
- Dereli-Öz, A.; Versini, G.; Halazonetis, T.D. Studies of genomic copy number changes in human cancers reveal signatures of DNA replication stress. Mol. Oncol. 2011, 5, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Herkert, B.; Eilers, M. Facilitating replication under stress: An oncogenic function of MYC? Nat. Rev. Cancer 2009, 9, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.; Herrador, R.; Lopes, M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.; Eder, A.; Mao, M.; Bast, R.C.; Boyd, D.; Mills, G.B. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 1999, 18, 6635–6640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, K.; Rak, J.; Jin, J.; Kerbel, R.S.; Newman, M.J.; Filmus, J. Downregulation of the pro-apoptotic protein Bak is required for the ras-induced transformation of intestinal epithelial cells. Curr. Biol. 1998, 8, 1331–1334. [Google Scholar] [CrossRef]
- Zhao, J.; Kennedy, B.K.; Lawrence, B.D.; Barbie, D.A.; Matera, A.G.; Fletcher, J.A.; Harlow, E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev. 2000, 14, 2283–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooley, A.; Zelivianski, S.; Jeruss, J.S. Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines. Cell Cycle 2010, 9, 4900–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Dalton, S. Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb. Perspect. Med. 2013, 3, a014381. [Google Scholar] [CrossRef] [PubMed]
- Neiman, P.E.; Thomas, S.J.; Loring, G. Induction of apoptosis during normal and neoplastic B-cell development in the bursa of Fabricius. Proc. Natl. Acad. Sci. USA 1991, 88, 5857–5861. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Alevizopoulos, K.; Vlach, J.; Hennecke, S.; Amati, B. Cyclin E and c-Myc promote cell proliferation in the presence of p16INK4a and of hypophosphorylated retinoblastoma family proteins. EMBO J. 1997, 16, 5322–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is Senescence Reversible? Curr. Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Larsson, L.G. Cdk2: A key regulator of the senescence control function of Myc. Aging (Albany NY) 2010, 2, 244–250. [Google Scholar] [CrossRef] [PubMed]
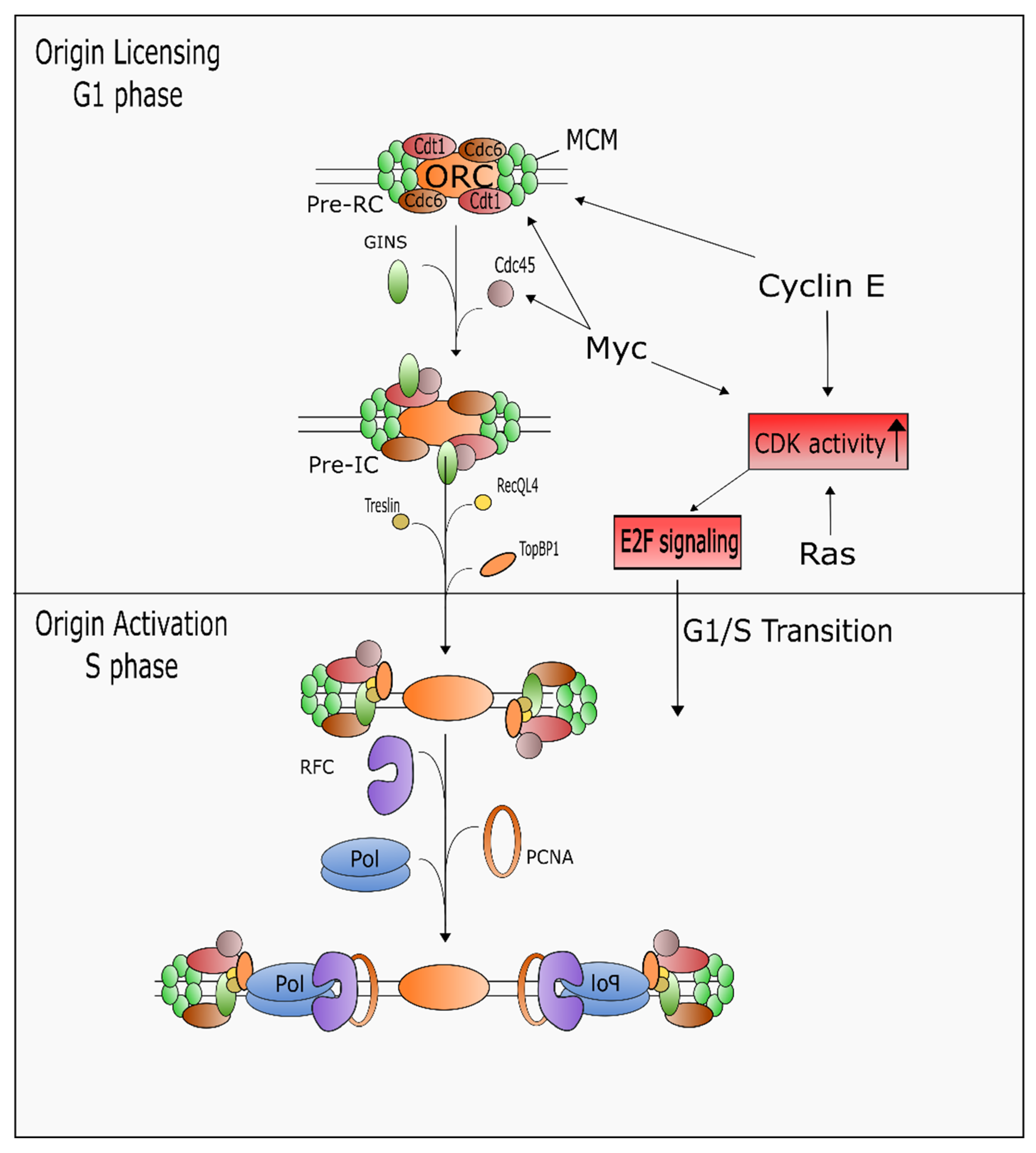
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Langston, L.D.; Mayle, R.; Schauer, G.D.; Yurieva, O.; Zhang, D.; Yao, N.Y.; Georgescu, R.E.; O’Donnell, M.E. Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks. Elife 2017, 6, e29118. [Google Scholar] [CrossRef] [PubMed]
- Ricke, R.M.; Bielinsky, A.K. Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Mol. Cell 2004, 16, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Cheng, I.H.; Chai, W.; Tye, B.K. Mcm10 and Cdc45 cooperate in origin activation in Saccharomyces cerevisiae. J. Mol. Biol. 2004, 340, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Langston, L.D.; Zhang, D.; Yurieva, O.; Georgescu, R.E.; Finkelstein, J.; Yao, N.Y.; Indiani, C.; O’Donnell, M.E. CMG helicase and DNA polymerase epsilon form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc. Natl. Acad. Sci. USA 2014, 111, 15390–15395. [Google Scholar] [CrossRef] [PubMed]
- Sanada, I.; Nakada, K.; Furugen, S.; Kumagai, E.; Yamaguchi, K.; Yoshida, M.; Takatsuki, K. Chromosomal abnormalities in a patient with smoldering adult T-cell leukemia: Evidence for a multistep pathogenesis. Leuk. Res. 1986, 10, 1377–1382. [Google Scholar] [CrossRef]
- Borlado, L.R.; Mendez, J. CDC6: From DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis 2008, 29, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, Y.; Sugimoto, N.; Yugawa, T.; Narisawa-Saito, M.; Kiyono, T.; Fujita, M. Deregulation of Cdt1 induces chromosomal damage without rereplication and leads to chromosomal instability. J. Cell Sci. 2006, 119 Pt 15, 3128–3140. [Google Scholar] [CrossRef]
- Hills, S.A.; Diffley, J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Ye, S.; Wang, X.; Zhang, Y.; Yan, D.; Cai, X.; Gao, W.; Shan, H.; Gao, Y.; Chen, J.; et al. Pre-RC Protein MCM7 depletion promotes mitotic exit by Inhibiting CDK1 activity. Sci. Rep. 2017, 7, 2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekholm-Reed, S.; Méndez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Valovka, T.; Schönfeld, M.; Raffeiner, P.; Breuker, K.; Dunzendorfer-Matt, T.; Hartl, M.; Bister, K. Transcriptional control of DNA replication licensing by Myc. Sci. Rep. 2013, 3, 3444. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.J.; Wu, S.P.; Liu, J.B.; Shi, Y.S.; Huang, X.; Zhang, Q.B.; Yao, K.T. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2013, 73, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Moser, R.; Toyoshima, M.; Robinson, K.; Gurley, K.E.; Howie, H.L.; Davison, J.; Morgan, M.; Kemp, C.J.; Grandori, C. MYC-driven tumorigenesis is inhibited by WRN syndrome gene deficiency. Mol. Cancer Res. 2012, 10, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Lee, H.O.; Bunker, B.D.; Dorn, E.S.; Rogers, G.C.; Duronio, R.J.; Cook, J.G. Cdt1 and Cdc6 are destabilized by rereplication-induced DNA damage. J. Biol. Chem. 2008, 283, 25356–25363. [Google Scholar] [CrossRef] [PubMed]
- Davidson, I.F.; Li, A.; Blow, J.J. Deregulated replication licensing causes DNA fragmentation consistent with head-to-tail fork collision. Mol. Cell 2006, 24, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.; Dowdy, S.F. Regulation of G(1) cell-cycle progression by oncogenes and tumor suppressor genes. Curr. Opin. Genet. Dev. 2002, 12, 47–52. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C. Metabolic Dependencies in RAS-Driven Cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Lee, E.S.; You, H.J.; Lee, J.W.; Park, J.W.; Chun, Y.S. Ras-dependent induction of HIF-1α785 via the Raf/MEK/ERK pathway: A novel mechanism of Ras-mediated tumor promotion. Oncogene 2004, 23, 9427–9431. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Li, F.; Handler, J.; Huang, C.R.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelekar, A.; Cole, M.D. Immortalization by c-myc, H-ras, and Ela oncogenes induces differential cellular gene expression and growth factor responses. Mol. Cell. Biol. 1987, 7, 3899–3907. [Google Scholar] [CrossRef] [PubMed]
- Elenbaas, B.; Spirio, L.; Koerner, F.; Fleming, M.D.; Zimonjic, D.B.; Donaher, J.L.; Popescu, N.C.; Hahn, W.C.; Weinberg, R.A. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001, 15, 50–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, K.M.; Worth, A.J.; Snyder, N.W.; Lee, J.V.; Sivanand, S.; Liu, Q.; Blair, I.A.; Wellen, K.E.; Zhang, R. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 2015, 11, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, Y.; Mutter-Rottmayer, L.; Zlatanou, A.; Durando, M.; Ding, W.; Wyatt, D.; Ramsden, D.; Tanoue, Y.; Tateishi, S.; et al. DNA repair factor RAD18 and DNA polymerase Polκ confer tolerance of oncogenic DNA replication stress. J. Cell Biol. 2017, 216, 3097–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, T.M.; Sharma, S.; Chabes, A.; Shcherbakova, P.V. Colon cancer-associated mutator DNA polymerase δ variant causes expansion of dNTP pools increasing its own infidelity. Proc. Natl. Acad. Sci. USA 2015, 112, E2467–E2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, L.N.; Marjavaara, L.; Knowels, G.M.; Schultz, E.M.; Fox, E.J.; Chabes, A.; Herr, A.J. dNTP pool levels modulate mutator phenotypes of error-prone DNA polymerase ε variants. Proc. Natl. Acad. Sci. USA 2015, 112, E2457–E2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, C.K. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 2015, 15, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Moparthy, K.C.; Wheeler, L.J.; Natarajan, V.; Zucker, S.N.; Fink, E.E.; Im, M.; Flanagan, S.; Burhans, W.C.; Zeitouni, N.C.; et al. Depletion of deoxyribonucleotide pools is an endogenous source of DNA damage in cells undergoing oncogene-induced senescence. Am. J. Pathol. 2013, 182, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.T.; Reyes, G.; Gries, K.; Ceylan, C.; Sharma, S.; Meurer, M.; Knop, M.; Chabes, A.; Hombauer, H. Alterations in cellular metabolism triggered by. Proc. Natl. Acad. Sci. USA 2017, 114, E4442–E4451. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Disbrow, G.L.; Yuan, H.; Tomaic, V.; Schlegel, R. Myc and human papillomavirus type 16 E7 genes cooperate to immortalize human keratinocytes. J. Virol. 2007, 81, 12689–12695. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, K.; Sekimoto, T.; Oda, T.; Kawabata, T.; Hanaoka, F.; Yamashita, T. Polη, a Y-family translesion synthesis polymerase, promotes cellular tolerance of Myc-induced replication stress. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Hedglin, M.; Pandey, B.; Benkovic, S.J. Characterization of human translesion DNA synthesis across a UV-induced DNA lesion. Elife 2016, 5, e19788. [Google Scholar] [CrossRef] [PubMed]
- Wittig, B.; Wölfl, S.; Dorbic, T.; Vahrson, W.; Rich, A. Transcription of human c-myc in permeabilized nuclei is associated with formation of Z-DNA in three discrete regions of the gene. EMBO J. 1992, 11, 4653–4663. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; De Silva, E.; Tornaletti, S.; Wang, G.; Vasquez, K.M.; Hanawalt, P.C. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J. Biol. Chem. 2007, 282, 32433–32441. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Mundo, I.M.A.; Zewail-Foote, M.; Kerwin, S.M.; Vasquez, K.M. Alternative DNA structure formation in the mutagenic human c-MYC promoter. Nucleic Acids Res. 2017, 45, 4929–4943. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.P.; Tsao, W.; Moldovan, G.; Eckert, K. DNA Polymerase Eta Prevents Tumor Cell Cycle Arrest and Cell Death During Recovery from Replication Stress. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Yan, Y.; Gerson, S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair (Amst) 2018, 66–67, 24–29. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsao, W.-C.; Eckert, K.A. Detours to Replication: Functions of Specialized DNA Polymerases during Oncogene-induced Replication Stress. Int. J. Mol. Sci. 2018, 19, 3255. https://doi.org/10.3390/ijms19103255
Tsao W-C, Eckert KA. Detours to Replication: Functions of Specialized DNA Polymerases during Oncogene-induced Replication Stress. International Journal of Molecular Sciences. 2018; 19(10):3255. https://doi.org/10.3390/ijms19103255
Chicago/Turabian StyleTsao, Wei-Chung, and Kristin A. Eckert. 2018. "Detours to Replication: Functions of Specialized DNA Polymerases during Oncogene-induced Replication Stress" International Journal of Molecular Sciences 19, no. 10: 3255. https://doi.org/10.3390/ijms19103255
APA StyleTsao, W. -C., & Eckert, K. A. (2018). Detours to Replication: Functions of Specialized DNA Polymerases during Oncogene-induced Replication Stress. International Journal of Molecular Sciences, 19(10), 3255. https://doi.org/10.3390/ijms19103255