PR/SET Domain Family and Cancer: Novel Insights from The Cancer Genome Atlas


 ,
,  , ,
, , 
 ,
,  and
and
Abstract
:
1. Introduction
2. Results
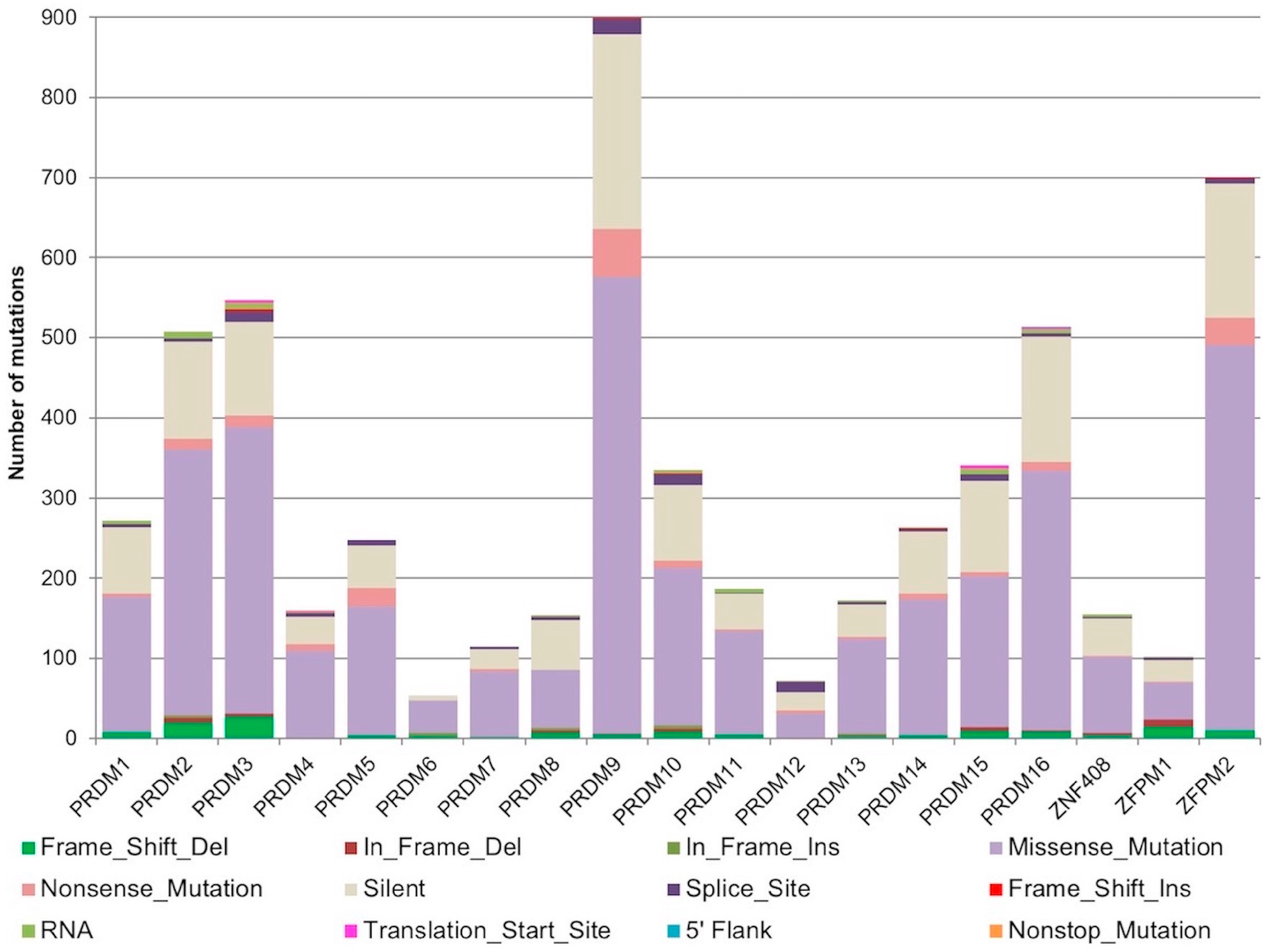
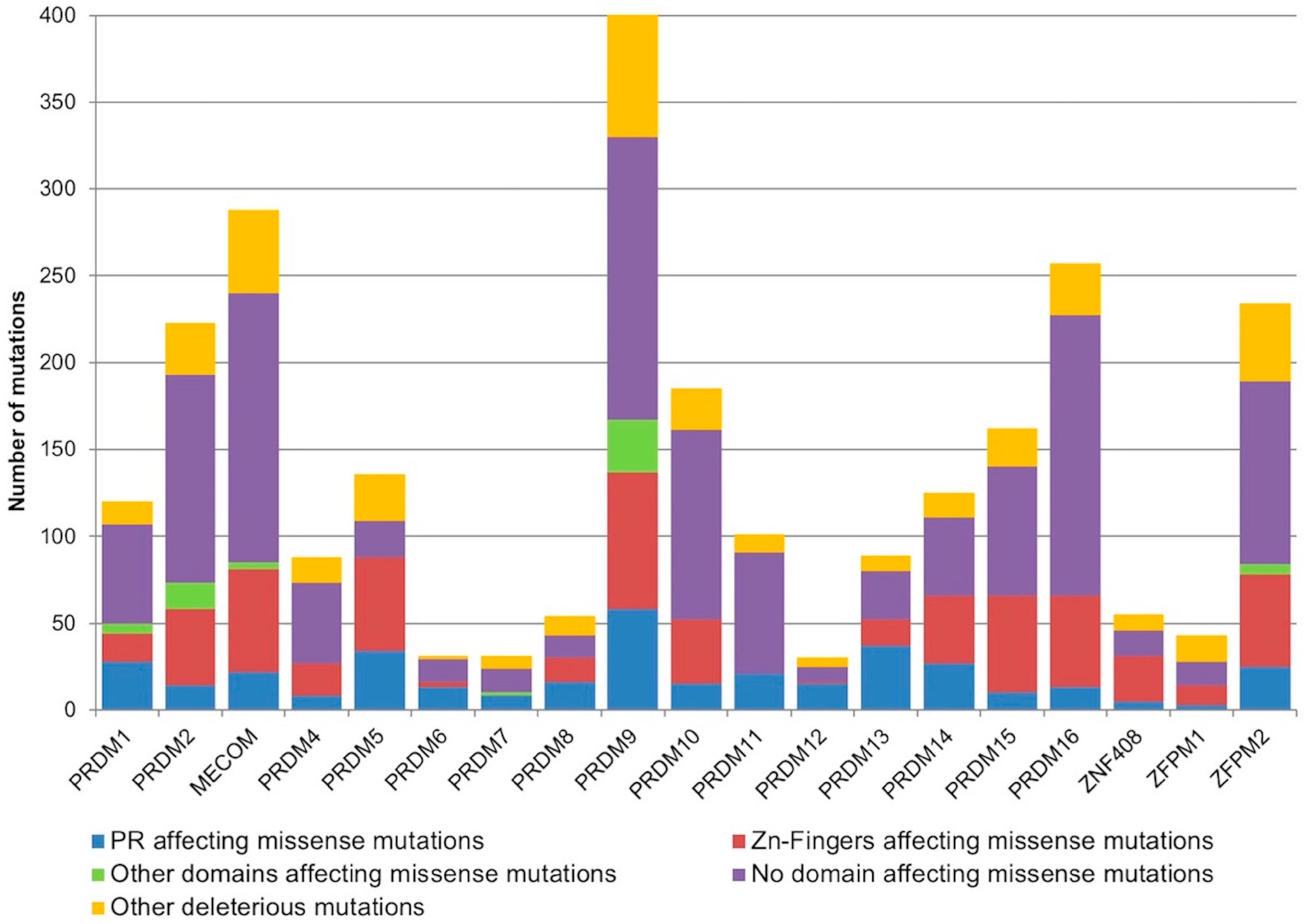
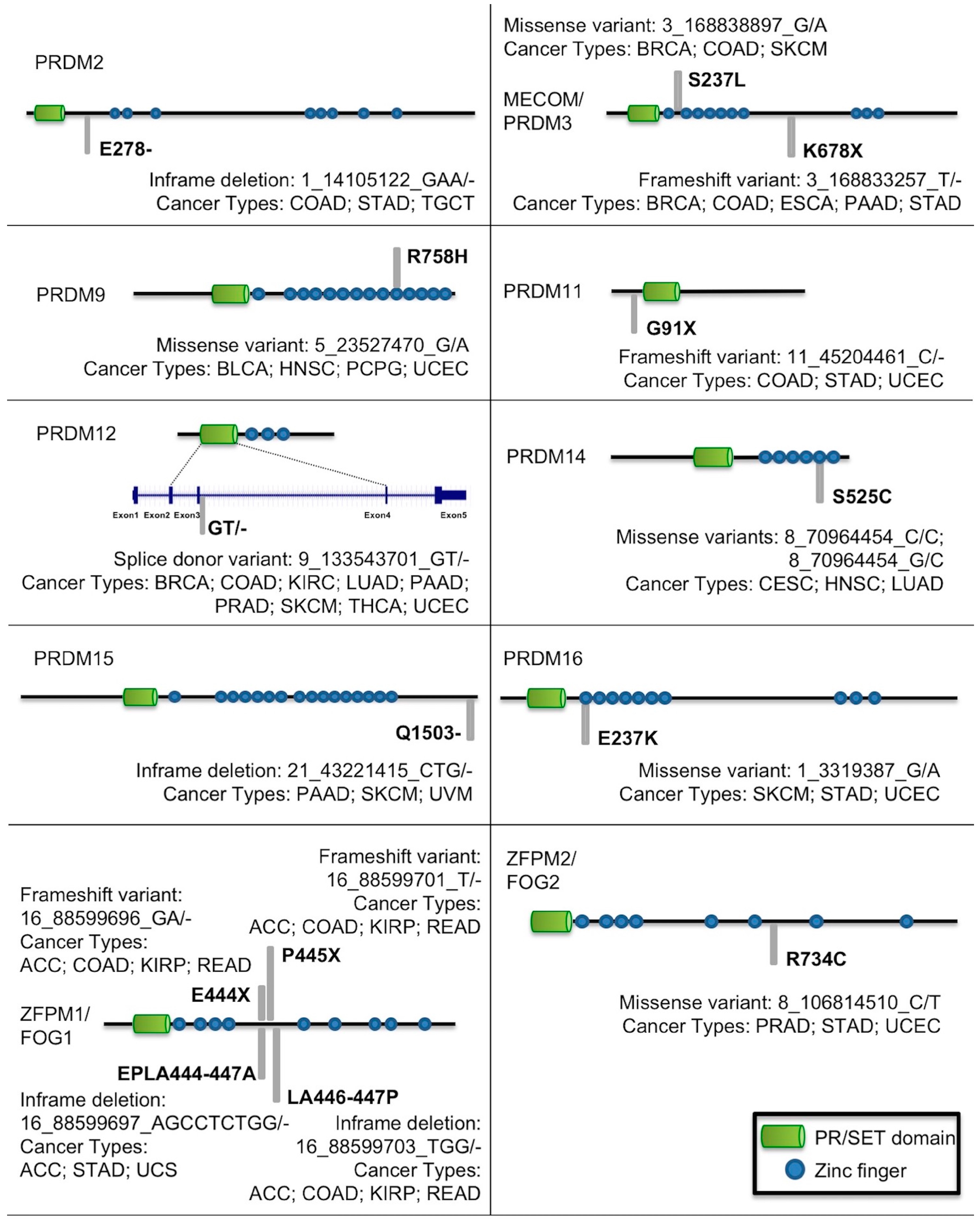
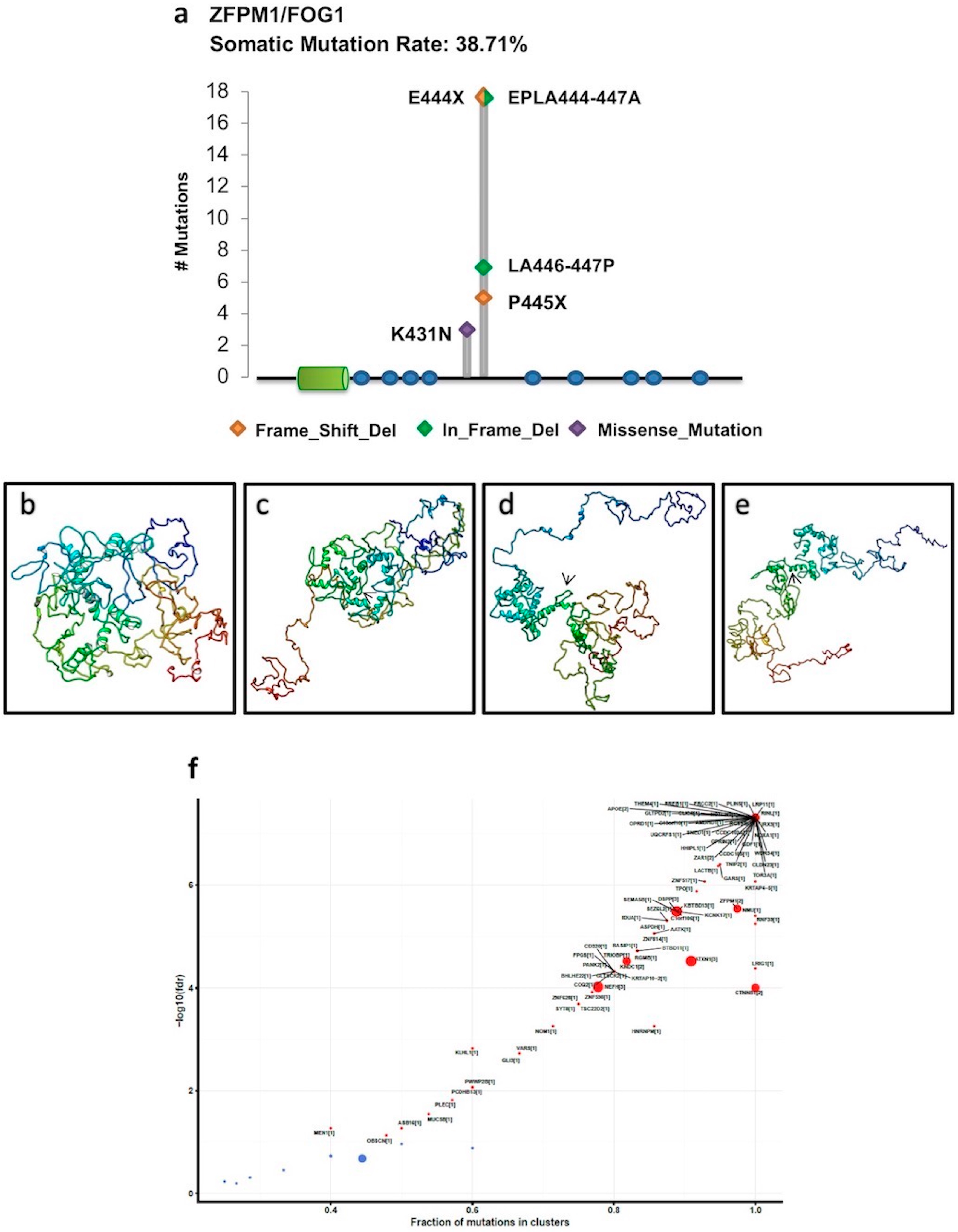
2.1. Mutational Profiling of PRDM Genes Across Human Cancers
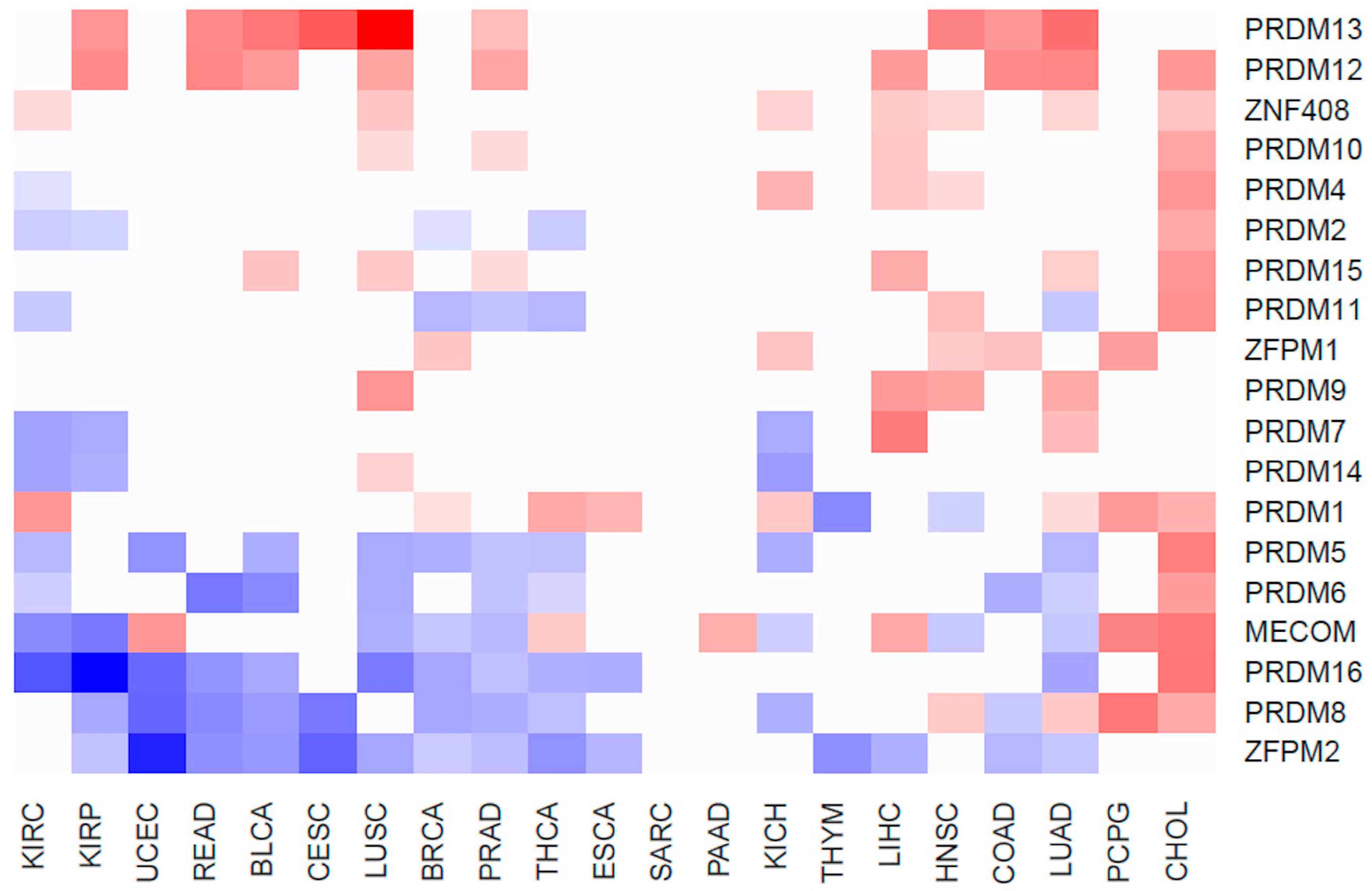
2.2. Differentially Expressed PRDM Genes across Human Cancers
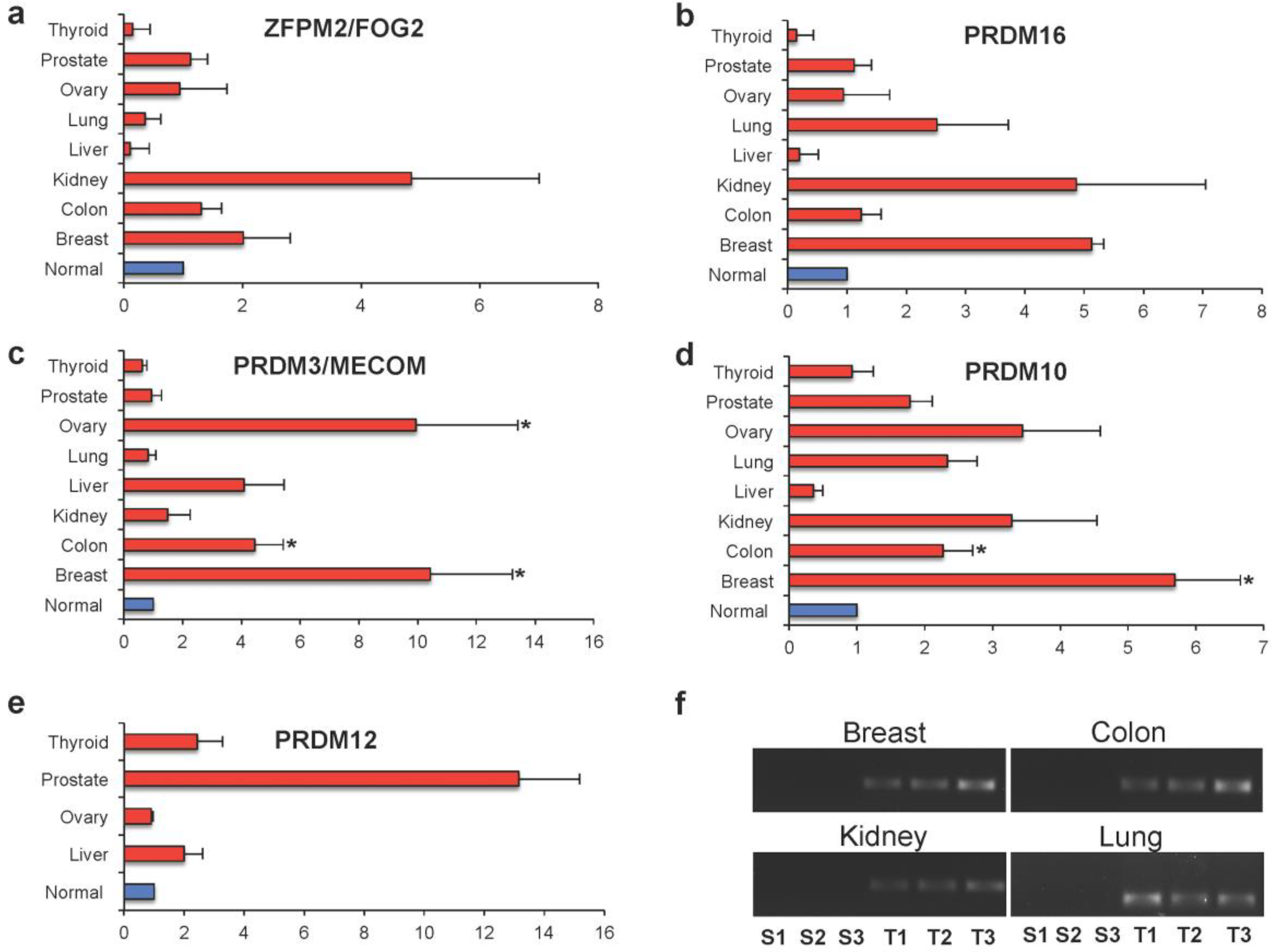
2.3. PRDM Expression in Human Primary Tumors
3. Discussion
4. Materials and Methods
4.1. TCGA Data Source Selection and Processing for Mutation Analysis
4.2. TCGA Data Source Selection and Processing for Expression Analysis
4.3. Real-Time RT-PCR Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FC | Fold change |
| FDR | False discovery rate |
| TCGA | The Cancer Genome Atlas |
| ACC | Adrenocortical carcinoma |
| BLCA | Bladder cancer |
| BRCA | Breast cancer |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma |
| CHOL | Cholangiocarcinoma |
| COAD | Colon adenocarcinoma |
| DLBC | Lymphoid neoplasm diffuse large B-cell lymphoma |
| ESCA | Esophageal carcinoma |
| GBM | Glioblastoma |
| HNSC | Head and neck squamous cell carcinoma |
| KICH | Kidney chromophobe carcinoma |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LAML | Acute myeloid leukemia |
| LIHC | Liver hepatocarcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| OV | Ovarian cancer |
| PAAD | Pancreas adenocarcinoma |
| PCPG | Pheochromocytoma and paraganglioma |
| PRAD | Prostate adenocarcinoma |
| READ | Rectum adenocarcinoma |
| SARC | Sarcoma |
| SKCM | Skin cutaneous melanoma |
| STAD | Stomach adenocarcinoma |
| TGCT | Testicular germ cell tumors |
| THCA | Thyroid cancer |
| THYM | Thymoma |
| UCEC | Uterine corpus endometrial carcinoma |
| UCS | Uterine carcinosarcoma |
| UVM | Uveal melanoma |
References
- Fumasoni, I.; Meani, N.; Rambaldi, D.; Scafetta, G.; Alcalay, M.; Ciccarelli, F.D. Family expansion and gene rearrangements contributed to the functional specialization of PRDM genes in vertebrates. BMC. Evol. Biol. 2007, 7, 187. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Xu, P.F.; Zhou, T.; Hu, M.; Fu, C.T.; Zhang, Y.; Jin, Y.; Chen, Y.; Chen, S.J.; Huang, Q.H.; et al. Genome-wide survey and developmental expression mapping of zebrafish SET domain-containing genes. PLoS ONE 2008, 3, e1499. [Google Scholar] [CrossRef] [PubMed]
- Clifton, M.K.; Westman, B.J.; Thong, S.Y.; O’Connell, M.R.; Webster, M.W.; Shepherd, N.E.; Quinlan, K.G.; Crossley, M.; Blobel, G.A.; Mackay, J.P. The identification and structure of an N-terminal PR domain show that FOG1 is a member of the PRDM family of proteins. PLoS ONE 2014, 9, e106011. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, M.; Meulemeester, D.; Béhague, J.; Kerner, P. Evolution of Prdm Genes in Animals: Insights from Comparative Genomics. Mol. Biol. Evol. 2016, 33, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Hohenauer, T.; Moore, A.W. The Prdm family: Expanding roles in stem cells and development. Development 2012, 139, 2267–2282. [Google Scholar] [CrossRef] [PubMed]
- Fog, C.K.; Galli, G.G.; Lund, A.H. PRDM proteins: Important players in differentiation and disease. BioEssays 2012, 34, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; De Rosa, C.; Abbondanza, C.; Moncharmont, B. PRDM Proteins: Molecular Mechanisms in Signal Transduction and Transcriptional Regulation. Biology 2013, 2, 107–141. [Google Scholar] [CrossRef] [PubMed]
- Mzoughi, S.; Tan, Y.X.; Low, D.; Guccione, E. The role of PRDMs in cancer: One family, two sides. Curr. Opin. Genet. Dev. 2016, 36, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Shao, G.; Liu, L. The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J. Biol. Chem. 1998, 273, 15933–15939. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Chee, K.J.; Kim, T.H.; Maniatis, T. PRDI-BF1/Blimp-1 repression is mediated by corepressors of the Groucho family of proteins. Genes Dev. 1999, 13, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajimura, S.; Seale, P.; Kubota, K.; Lunsford, E.; Frangioni, J.V.; Gygi, S.P.; Spiegelman, B.M. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 2009, 460, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okashita, N.; Suwa, Y.; Nishimura, O.; Sakashita, N.; Kadota, M.; Nagamatsu, G.; Kawaguchi, M.; Kashida, H.; Nakajima, A.; Tachibana, M.; et al. PRDM14 Drives OCT3/4 Recruitment via Active Demethylation in the Transition from Primed to Naive Pluripotency. Stem Cell Rep. 2016, 7, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Cohen, P. The Multifaceted Roles of PRDM16: Adipose Biology and Beyond. Trends Endocrinol. Metab. 2016, 27, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Rienzo, M.; Ciccodicola, A.; Casamassimi, A.; Abbondanza, C. Human PRDM2: Structure, function and pathophysiology. Biochim. Biophys. Acta 2018, 1861, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Maruvka, Y.E.; Mouw, K.W.; Karlic, R.; Parasuraman, P.; Kamburov, A.; Polak, P.; Haradhvala, N.J.; Hess, J.M.; Rheinbay, E.; Brody, Y.; et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat. Biotechnol. 2017, 35, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Kim, M.S.; Song, S.Y.; Yoo, N.J.; Lee, S.H. Intratumoral Heterogeneity of Frameshift Mutations in MECOM Gene is Frequent in Colorectal Cancers with High Microsatellite Instability. Pathol. Oncol. Res. 2017, 23, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.H.; Hakim, O.; et al. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014, 8, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhu, Q.; He, H.; Jiang, L.; Qiang, Q.; Hu, L.; Hu, G.; Jiang, Y.; Ding, X.; Lu, Y. RIZ1: A potential tumor suppressor in glioma. BMC Cancer 2015, 15, 990. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zelazowski, M.J.; Cole, F. Missing the Mark: PRDM9-Dependent Methylation Is Required for Meiotic DSB Targeting. Mol. Cell 2018, 69, 725–727. [Google Scholar] [CrossRef]
- Diagouraga, B.; Clément, J.A.J.; Duret, L.; Kadlec, J.; de Massy, B.; Baudat, F. PRDM9 Methyltransferase Activity Is Essential for Meiotic DNA Double-Strand Break Formation at Its Binding Sites. Mol. Cell 2018, 69, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Geng, H.; Li, L.; Ying, J.; Ma, C.; Wang, Y.; Poon, F.F.; Wang, X.; Ying, Y.; Yeo, W.; et al. The Epigenetic Modifier PRDM5 Functions as a Tumor Suppressor through Modulating WNT/β-Catenin Signaling and Is Frequently Silenced in Multiple Tumors. PLoS ONE 2011, 6, e27346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meani, N.; Pezzimenti, F.; Deflorian, G.; Mione, M.; Alcalay, M. The tumor suppressor PRDM5 regulates Wnt signaling at early stages of zebrafish development. PLoS ONE 2009, 4, e4273. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, A.; Rienzo, M.; Abbondanza, C.; Costa, V.; Ciccodicola, A.; Casamassimi, A. Pan-Cancer Mutational and Transcriptional Analysis of the Integrator Complex. Int. J. Mol. Sci. 2017, 18, 936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Cancer genomes: Discerning drivers from passengers. Nat. Methods 2014, 11, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Kamburov, A.; Tamborero, D.; Pons, T.; Grases, D.; Valencia, A.; Lopez-Bigas, N.; Getz, G.; Godzik, A. Comparison of algorithms for the detection of cancer drivers at subgene resolution. Nat. Methods 2017, 14, 782–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Giner, F.; Ratcliffe, P.; Tomlinson, I. The mini-driver model of polygenic cancer evolution. Nat. Rev. Cancer 2015, 15, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Tsang, A.P.; Visvader, J.E.; Turner, C.A.; Fujiwara, Y.; Yu, C.; Weiss, M.J.; Crossley, M.; Orkin, S.H. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell 1997, 90, 109–119. [Google Scholar] [CrossRef]
- Cai, Q.; Jeannet, R.; Hua, W.K.; Cook, G.J.; Zhang, B.; Qi, J.; Liu, H.; Li, L.; Chen, C.C.; Marcucci, G.; et al. CBFβ-SMMHC creates aberrant megakaryocyte-erythroid progenitors prone to leukemia initiation in mice. Blood 2016, 128, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Ruffalo, M.; Koyutürk, M.; Sharan, R. Network-Based Integration of Disparate Omic Data to Identify “Silent Players” in Cancer. PLoS Comput. Biol. 2015, 11, e1004595. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chaudhary, K.; Garmire, L.X. More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front. Genet. 2017, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Koeffler, H.P. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. bioRxiv 2016. [Google Scholar] [CrossRef]
- HGNC. Available online: http://www.genenames.org (accessed on 19 October 2018).
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- TCGA. Available online: https://tcga-data.nci.nih.gov/tcga/ (accessed on 25 September 2017).
- Primer3Plus. Available online: http://primer3plus.com/cgi-bin/dev/primer3plus.cgi (accessed on 12 October 2017).
- UCSC-Genome Browser. Available online: https://genome.ucsc.edu (accessed on 8 January 2018).
- Rienzo, M.; Schiano, C.; Casamassimi, A.; Grimaldi, V.; Infante, T.; Napoli, C. Identification of valid reference housekeeping genes for gene expression analysis in tumor neovascularization studies. Clin. Transl. Oncol. 2013, 15, 211–218. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancers | Genes | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRDM1 | PRDM2 | MECOM | PRDM4 | PRDM5 | PRDM6 | PRDM7 | PRDM8 | PRDM9 | PRDM10 | PRDM11 | PRDM12 | PRDM13 | PRDM14 | PRDM15 | PRDM16 | ZNF408 | ZFPM1 | ZFPM2 | |
| ACC | 0 | 1.1 | 0 | 1.1 | 1.1 | 2.2 | 0 | 0 | 3.2 | 1.1 | 0 | 0 | 1.1 | 0 | 0 | 1.1 | 0 | 50.5 | 4.3 |
| BLCA | 1.7 | 4.1 | 5.3 | 3.4 | 3.4 | 0 | 1.9 | 1 | 3.9 | 2.2 | 1.7 | 0 | 1.4 | 1.7 | 2.7 | 4.1 | 0.7 | 1 | 3.6 |
| BRCA | 0.8 | 0.9 | 1 | 0.5 | 0.3 | 0.2 | 0.2 | 0.5 | 0.7 | 0.8 | 0.2 | 0.2 | 0.3 | 0.5 | 0.6 | 0.5 | 0.1 | 0.2 | 1.3 |
| CESC | 0.5 | 4 | 2 | 1.5 | 2 | 0.5 | 1 | 0.5 | 4.5 | 1 | 1.5 | 0 | 0 | 0.5 | 3 | 3 | 1 | 0.5 | 1 |
| CHOL | 2.7 | 0 | 0 | 5.4 | 0 | 2.7 | 0 | 0 | 5.4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.7 | 0 | 0 |
| COAD | 2.2 | 6.6 | 5.5 | 1.5 | 2.6 | 2.9 | 0.7 | 1.1 | 5.5 | 4.4 | 1.1 | 0.4 | 1.5 | 1.8 | 2.2 | 4.4 | 1.5 | 6.6 | 2.6 |
| DLBC | 8.2 | 2 | 2 | 0 | 2 | 0 | 0 | 2 | 8.2 | 0 | 0 | 0 | 0 | 0 | 2 | 6.1 | 0 | 0 | 4.1 |
| ESCA | 1.1 | 1.6 | 3.2 | 2.1 | 1.6 | 2.7 | 1.1 | 1.6 | 7 | 4.8 | 1.6 | 0 | 2.1 | 2.7 | 4.8 | 2.7 | 1.6 | 0.5 | 8.6 |
| GBM | 0.6 | 0.6 | 2.8 | 0.3 | 1.1 | 0 | 0.3 | 0 | 3.3 | 1.9 | 0.6 | 0.8 | 0.3 | 0.3 | 1.9 | 0.6 | 0.6 | 0 | 0.3 |
| HNSC | 1.3 | 2.1 | 2.5 | 0.8 | 1.3 | 0 | 0.6 | 0.6 | 7.2 | 1.1 | 1.1 | 0.8 | 0.8 | 1.9 | 0.9 | 2.3 | 0.6 | 0.9 | 0.8 |
| KICH | 0 | 1.5 | 1.5 | 0 | 0 | 0 | 0 | 1.5 | 6 | 0 | 0 | 1.5 | 1.5 | 0 | 0 | 1.5 | 0 | 0 | 0 |
| KIRC | 0.2 | 0.8 | 1.2 | 1.5 | 0.3 | 0.2 | 0.8 | 0 | 1 | 1 | 0.2 | 0.3 | 0.7 | 0.5 | 1.2 | 0.5 | 0.5 | 0.2 | 1.2 |
| KIRP | 0.3 | 2.4 | 0 | 0.3 | 0.3 | 0.7 | 0.3 | 0.3 | 0.7 | 1 | 0.7 | 0.3 | 1.4 | 1 | 1.7 | 1.4 | 1.4 | 0.3 | 0.3 |
| LAML | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 | 0 |
| LIHC | 1.6 | 3.2 | 2.9 | 1.8 | 1.1 | 0.8 | 0.5 | 0.8 | 2.4 | 1.6 | 1.8 | 0 | 0.8 | 1.3 | 2.4 | 2.1 | 0.8 | 0 | 3.9 |
| LUAD | 1.9 | 3.7 | 2.1 | 1.6 | 1.9 | 0 | 1.9 | 1.2 | 14.2 | 3 | 1.6 | 1.1 | 0.9 | 4.4 | 2.3 | 4.2 | 0.9 | 0.2 | 11.1 |
| LUSC | 2.8 | 3.4 | 5 | 0 | 2.8 | 0 | 0.6 | 0 | 7.3 | 3.4 | 2.2 | 1.1 | 3.4 | 0 | 2.2 | 3.4 | 1.7 | 0 | 5 |
| OV | 0.2 | 0.4 | 0 | 0 | 0 | 0 | 0.4 | 0 | 1.3 | 0 | 0.2 | 0 | 0 | 0.2 | 0.2 | 0 | 0 | 0 | 1.3 |
| PAAD | 0.5 | 4.3 | 2.7 | 1.1 | 1.6 | 0 | 2.1 | 16 | 1.6 | 3.2 | 2.7 | 2.7 | 1.6 | 0.5 | 11.2 | 1.6 | 1.1 | 0 | 2.7 |
| PCPG | 0 | 0.5 | 0 | 0 | 0 | 0 | 0 | 0 | 1.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0.5 |
| PRAD | 0 | 0.8 | 1.4 | 0 | 0.6 | 0 | 0.6 | 0.6 | 0.8 | 0.4 | 0.2 | 0.4 | 0.2 | 0.4 | 0.4 | 0.8 | 0.6 | 0.6 | 1.4 |
| READ | 0 | 2.6 | 1.7 | 2.6 | 3.4 | 0.9 | 0 | 0 | 2.6 | 1.7 | 0.9 | 0.9 | 0 | 0 | 1.7 | 0.9 | 0 | 9.4 | 5.1 |
| SARC | 1.1 | 1.9 | 1.9 | 1.1 | 0.8 | 2.7 | 0.8 | 0.4 | 3 | 1.5 | 1.1 | 0.4 | 0.8 | 1.5 | 3.4 | 3 | 0.8 | 0.8 | 2.3 |
| SKCM | 5.3 | 4.2 | 20.1 | 1.3 | 3.6 | 0 | 2.3 | 1.5 | 15.4 | 3.6 | 3 | 1.3 | 2.5 | 3.6 | 3.6 | 7.8 | 1.9 | 0.8 | 16.5 |
| STAD | 2.3 | 7.8 | 3.8 | 1 | 3 | 0 | 1 | 2.3 | 5.8 | 4.3 | 1.5 | 0.3 | 2.8 | 2.5 | 3 | 4.8 | 1.8 | 1 | 5.8 |
| TGCT | 0 | 2.5 | 0.6 | 0 | 0 | 1.3 | 0.6 | 1.9 | 2.5 | 2.5 | 0.6 | 0 | 1.3 | 0 | 0 | 1.3 | 0 | 1.3 | 0 |
| THCA | 0.4 | 0.6 | 0 | 0.4 | 0.2 | 0 | 0.8 | 0.4 | 1.2 | 0.8 | 0.2 | 0.2 | 0.2 | 0 | 0.4 | 0.8 | 0.4 | 0.4 | 0.4 |
| THYM | 2.4 | 3.2 | 3.2 | 2.4 | 0 | 2.4 | 3.2 | 0.8 | 3.2 | 1.6 | 0.8 | 0.8 | 0.8 | 2.4 | 2.4 | 3.2 | 0.8 | 0.8 | 1.6 |
| UCEC | 4 | 7.2 | 5.6 | 2.4 | 4.4 | 2 | 0.8 | 1.6 | 10 | 5.2 | 2.4 | 1.2 | 2.8 | 4.8 | 3.2 | 5.6 | 4 | 0.4 | 4.8 |
| UCS | 5.2 | 1.7 | 0 | 0 | 3.4 | 0 | 1.7 | 1.7 | 1.7 | 1.7 | 1.7 | 0 | 1.7 | 0 | 3.4 | 0 | 1.7 | 5.2 | 5.2 |
| UVM | 0 | 1.2 | 0 | 1.2 | 1.2 | 0 | 0 | 1.2 | 4.9 | 0 | 1.2 | 0 | 0 | 0 | 1.2 | 0 | 0 | 0 | 0 |
| Genes | Deleterious Mutations | Total Mutations | % Deleterious Mutations |
|---|---|---|---|
| PRDM1 | 120 | 272 | 44.1 |
| PRDM2 | 223 | 507 | 44.0 |
| MECOM/PRDM3 | 288 | 547 | 52.7 |
| PRDM4 | 88 | 160 | 55.0 |
| PRDM5 | 136 | 248 | 54.8 |
| PRDM6 | 31 | 53 | 58.5 |
| PRDM7 | 31 | 114 | 27.2 |
| PRDM8 | 54 | 154 | 35.1 |
| PRDM9 | 403 | 899 | 44.8 |
| PRDM10 | 185 | 335 | 55.2 |
| PRDM11 | 101 | 187 | 54.0 |
| PRDM12 | 30 | 72 | 41.7 |
| PRDM13 | 89 | 172 | 51.7 |
| PRDM14 | 125 | 263 | 47.5 |
| PRDM15 | 162 | 341 | 47.5 |
| PRDM16 | 257 | 514 | 50.0 |
| ZNF408/PRDM17 | 55 | 155 | 35.5 |
| ZFPM1/FOG1 | 43 | 102 | 42.2 |
| ZFPM2/FOG2 | 234 | 700 | 33.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorrentino, A.; Federico, A.; Rienzo, M.; Gazzerro, P.; Bifulco, M.; Ciccodicola, A.; Casamassimi, A.; Abbondanza, C. PR/SET Domain Family and Cancer: Novel Insights from The Cancer Genome Atlas. Int. J. Mol. Sci. 2018, 19, 3250. https://doi.org/10.3390/ijms19103250
Sorrentino A, Federico A, Rienzo M, Gazzerro P, Bifulco M, Ciccodicola A, Casamassimi A, Abbondanza C. PR/SET Domain Family and Cancer: Novel Insights from The Cancer Genome Atlas. International Journal of Molecular Sciences. 2018; 19(10):3250. https://doi.org/10.3390/ijms19103250
Chicago/Turabian StyleSorrentino, Anna, Antonio Federico, Monica Rienzo, Patrizia Gazzerro, Maurizio Bifulco, Alfredo Ciccodicola, Amelia Casamassimi, and Ciro Abbondanza. 2018. "PR/SET Domain Family and Cancer: Novel Insights from The Cancer Genome Atlas" International Journal of Molecular Sciences 19, no. 10: 3250. https://doi.org/10.3390/ijms19103250
APA StyleSorrentino, A., Federico, A., Rienzo, M., Gazzerro, P., Bifulco, M., Ciccodicola, A., Casamassimi, A., & Abbondanza, C. (2018). PR/SET Domain Family and Cancer: Novel Insights from The Cancer Genome Atlas. International Journal of Molecular Sciences, 19(10), 3250. https://doi.org/10.3390/ijms19103250