The Optimal Radiation Dose to Induce Robust Systemic Anti-Tumor Immunity


Abstract
:1. Introduction
2. Results
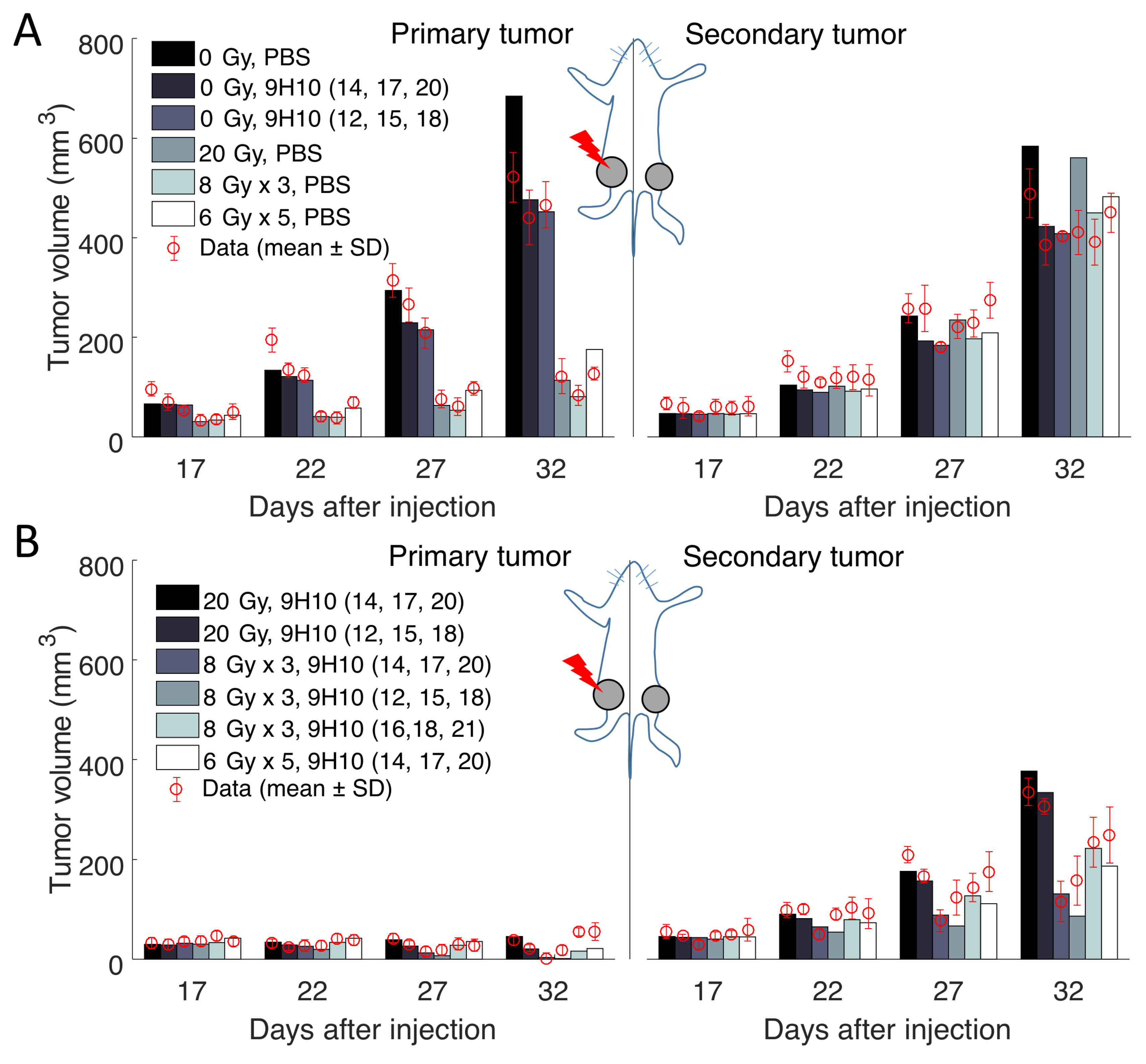
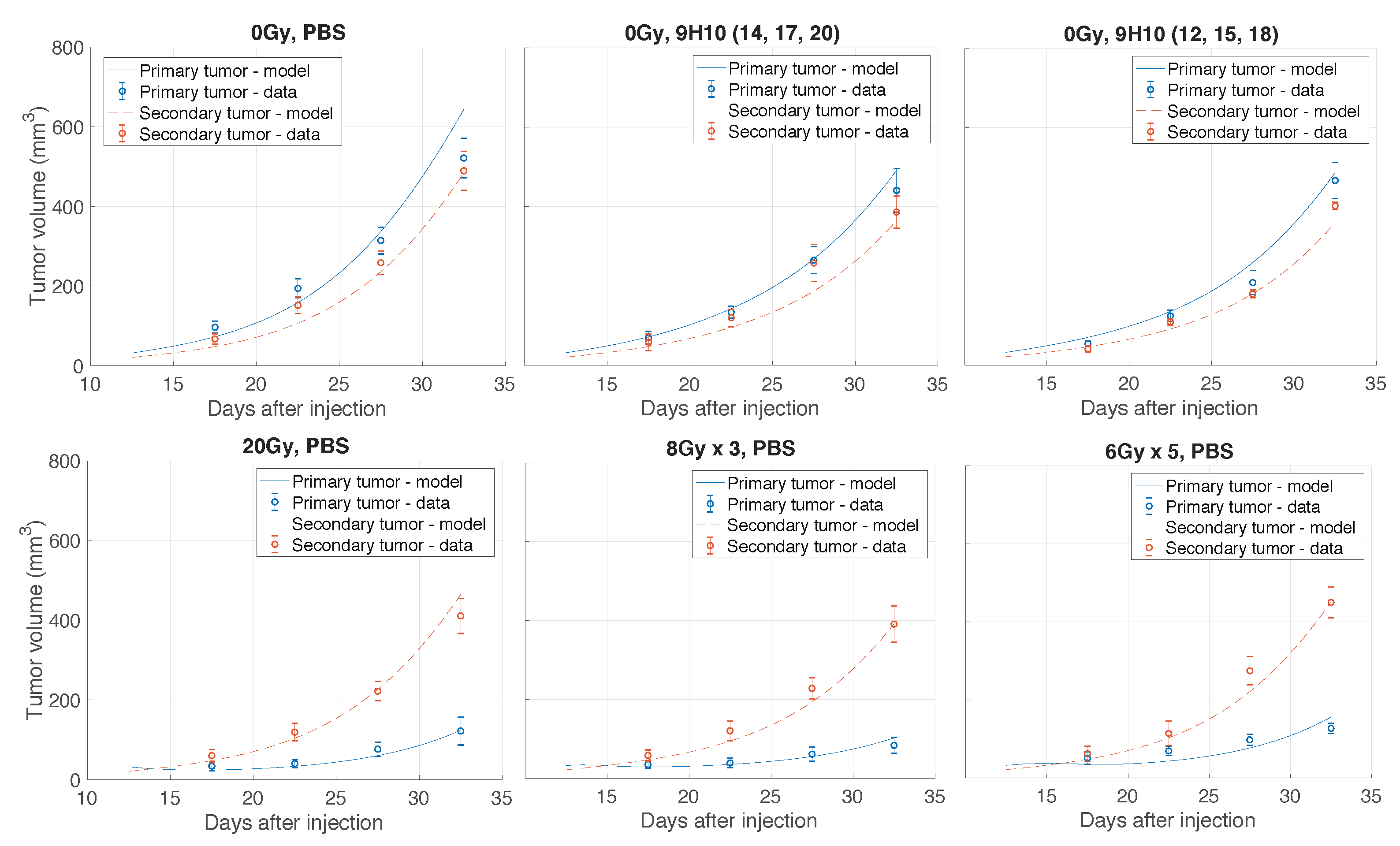
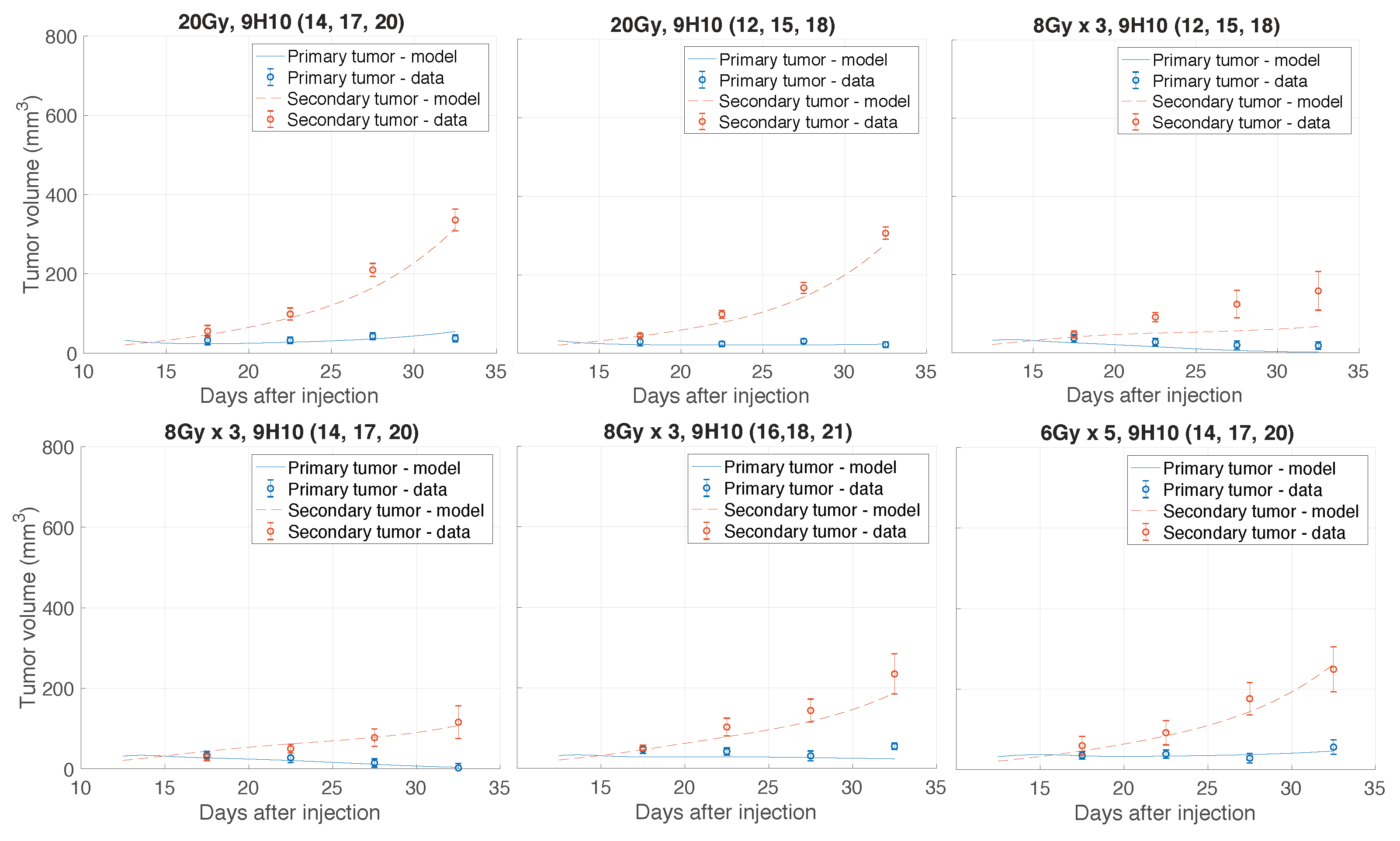
2.1. Data Fitting
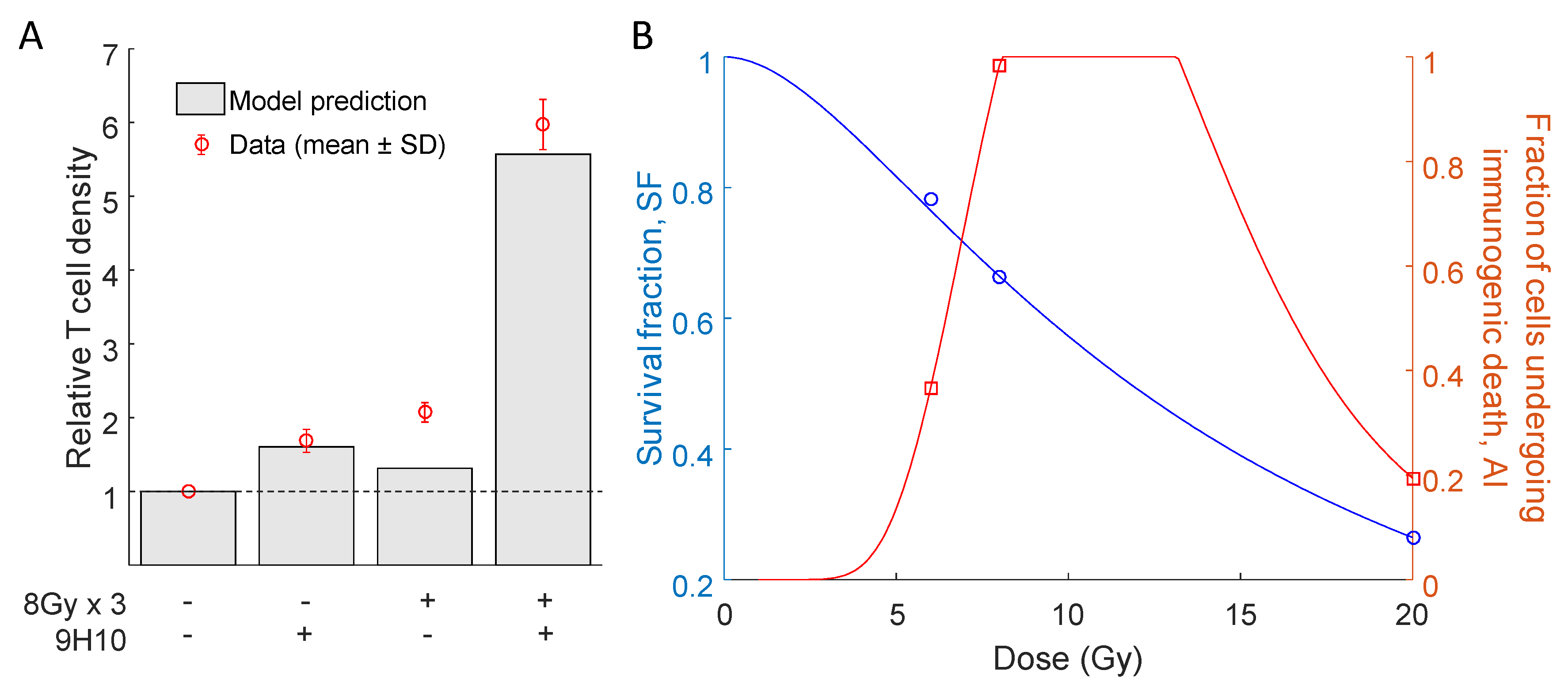
2.2. Predicted Radiation Response
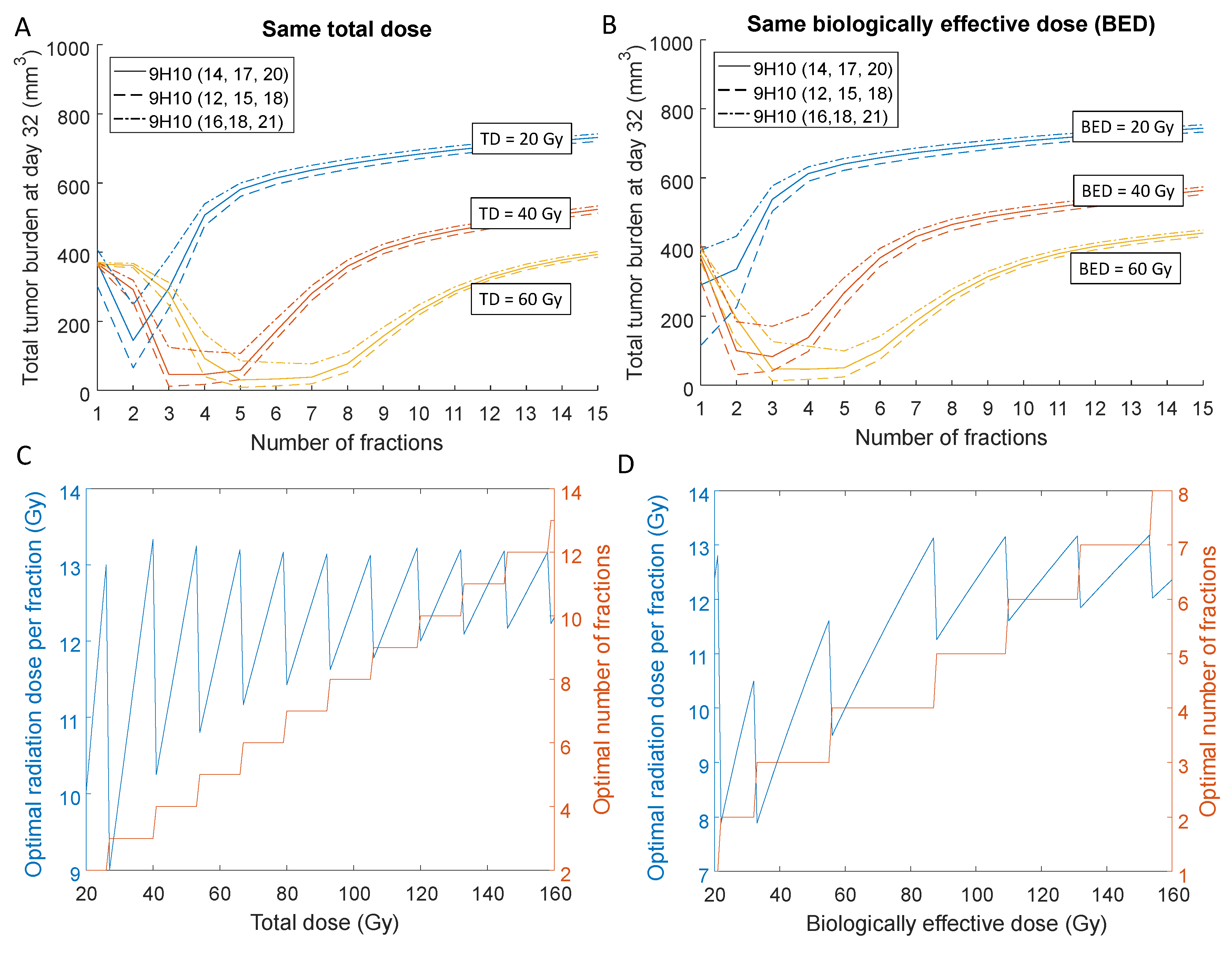
2.3. Optimal Radiation Dose and Dose Fractionation
3. Discussion
4. Materials and Methods
4.1. Experimental Data
4.2. Mathematical Model of Tumors-Immune System Interaction
4.3. Parameter Estimation
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RT | Radiotherapy |
| APC | Antigen-presenting cell |
| CTL | Cytotoxic T lymphocyte |
| IR | Irradiation |
| BED | Biologically effective dose |
Appendix A. Supplementary Data


References
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Erridge, S.C.; Featherstone, C.; Chalmers, R.; Campbell, J.; Stockton, D.; Black, R. What will be the radiotherapy machine capacity required for optimal delivery of radiotherapy in Scotland in 2015? Eur. J. Cancer 2007, 43, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Wideł, M.; Lalik, A.; Krzywoń, A.; Poleszczuk, J.; Fujarewicz, K.; Rzeszowska-Wolny, J. The different radiation response and radiation-induced bystander effects in colorectal carcinoma cells differing in p53 status. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2015, 778, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Poleszczuk, J.; Krzywoń, A.; Foryś, U.; Wideł, M. Connecting radiation-induced bystander effects and senescence to improve radiation response prediction. Radiat. Res. 2015, 183, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Blyth, B.J.; Sykes, P.J. Radiation-Induced Bystander Effects: What Are They, and How Relevant Are They to Human Radiation Exposures? Radiat. Res. 2011, 176, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Poleszczuk, J.; Luddy, K.A.; Prokopiou, S.; Robertson-Tessi, M.; Moros, E.G.; Fishman, M.; Djeu, J.Y.; Finkelstein, S.E.; Enderling, H. Abscopal benefits of localized radiotherapy depend on activated T cell trafficing and distribution between metastatic lesions. Cancer Res. 2016, 76, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Chhabra, A.; Chachoua, A.; Adams, S.; Donach, M.; Fenton-Kerimian, M.; Friedman, K.; Ponzo, F.; Babb, J.S.; Goldberg, J.; et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: A proof-of-principle trial. Lancet Oncol. 2015, 16, 795–803. [Google Scholar] [CrossRef]
- Crittenden, M.; Kohrt, H.; Levy, R.; Jones, J.; Camphausen, K.; Dicker, A.; Demaria, S.; Formenti, S. Current clinical trials testing combinations of immunotherapy and radiation. Semin. Radiat. Oncol. 2015, 25, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, J.M.; Shinohara, E.; Summers, J.B.; Niermann, K.J.; Morimoto, A.; Brousal, J. The controversial abscopal effect. Cancer Treat. Rev. 2005, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.A.; Wilhite, T.J.; Balboni, T.A.; Alexander, B.M.; Spektor, A.; Ott, P.A.; Ng, A.K.; Hodi, F.S.; Schoenfeld, J.D. A systematic evaluation of abscopal responses following radiotherapy in patients with metastatic melanoma treated with ipilimumab. Oncoimmunology 2015, 4, e1046028. [Google Scholar] [CrossRef] [PubMed]
- Poleszczuk, J.; Luddy, K.; Chen, L.; Lee, J.K.; Harrison, L.B.; Czerniecki, B.J.; Soliman, H.; Enderling, H. Neoadjuvant radiotherapy of early-stage breast cancer and long-term disease-free survival. Breast Cancer Res. 2017, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Pilones, K.A.; Kawashima, N.; Yang, A.M.; Babb, J.S.; Formenti, S.C.; Demaria, S. Invariant natural killer T cells regulate breast cancer response to radiation and CTLA-4 blockade. Clin. Cancer Res. 2009, 15, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Habets, T.H.; Oth, T.; Houben, A.W.; Huijskens, M.J.; Senden-Gijsbers, B.L.; Schnijderberg, M.C.; Brans, B.; Dubois, L.J.; Lambin, P.; De Saint-Hubert, M.; et al. Fractionated radiotherapy with 3 × 8 Gy induces systemic anti-tumour responses and abscopal tumour inhibition without modulating the humoral anti-tumour response. PLoS ONE 2016, 11, e0159515. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful anti-tumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Vatner, R.E.; Cooper, B.T.; Vanpouille-Box, C.; Demaria, S.; Formenti, S.C. Combinations of immunotherapy and radiation in cancer therapy. Front. Oncol. 2014, 4, 325. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2014, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.K.; Wolchok, J.D.; Allison, J.P. AntiCTLA-4 antibody therapy: Immune monitoring during clinical development of a novel immunotherapy. Semin. Oncol. 2010, 37, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Saenger, Y. The Mechanism of Anti-CTLA-4 Activity and the Negative Regulation of T-Cell Activation. Oncologist 2008, 13, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enderling, H.; Kim, S.; Pilon-Thomas, S. The Accelerating Quest For Optimal Radiation And Immunotherapy Combinations For Local And Systemic Tumor Control. Ther. Radiol. Oncol. 2018, 2, 33. [Google Scholar] [CrossRef]
- Walker, R.; Enderling, H. From concept to clinic: Mathematically informed immunotherapy. Curr. Probl. Cancer 2016, 40, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Sachs, R.K.; Hahnfeld, P.; Brenner, D.J. The link between low-LET dose-response relations and the underlying kinetics of damage production/repair/misrepair. Int. J. Radiat. Biol. 1997, 72, 351–374. [Google Scholar] [PubMed]
- Prokopiou, S.; Moros, E.G.; Poleszczuk, J.; Caudell, J.; Torres-Roca, J.F.; Latifi, K.; Lee, J.K.; Myerson, R.; Harrison, L.B.; Enderling, H. A proliferation saturation index to predict radiation response and personalize radiotherapy fractionation. Radiat. Oncol. 2015, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Poleszczuk, J.; Walker, R.; Moros, E.G.; Latifi, K.; Caudell, J.J.; Enderling, H. Predicting Patient-Specific Radiotherapy Protocols Based on Mathematical Model Choice for Proliferation Saturation Index. Bull. Math. Biol. 2018, 80, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Niedermann, G. Abscopal Effects With Hypofractionated Schedules Extending Into The Effector Phase Of The Tumor-Specific T-Cell Response. Int. J. Radiat. Onclol. Biol. Phys. 2018, 101, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.; Poleszczuk, J.; Pilon-Thomas, S.; Anderson, A.; Czerniecki, B.; Harrison, L.; Moros, E.; Enderling, H. Immune interconnectivity of anatomically distant tumors as a potential mediator of systemic responses to local therapy. Sci. Rep. 2018, 8, 9474. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.A.; Makalkin, I.A.; Taylor, M.A.; Perelson, A.S. Nonlinear dynamics of immunogenic tumors: Parameter estimation and global bifurcation analysis. Bull. Math. Biol. 1994, 56, 295–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, V.A.; Knott, G.D. Modeling tumor regrowth and immunotherapy. Math. Comput. Model. 2001, 33, 1275–1287. [Google Scholar] [CrossRef]
- Bonate, P.L. Pharmacokinetic-Pharmacodynamic Modeling and Simulation; Springer: Boston, MA, USA, 2011. [Google Scholar]
- Kirchner, D.; Panetta, J.C. Modeling immunotherapy of the tumor-immune interaction. J. Math. Biol. 1998, 37, 235–252. [Google Scholar] [CrossRef]
- Byrd, R.H.; Schnabel, R.B.; Schultz, G.A. A trust region algorithm for nonlinearly constrained optimization. SIAM J. Numer. Anal. 1987, 24, 1151–1170. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Unit | Value |
|---|---|---|---|
| r | Viable cancer cells volume doubling time | 1/day | 0.195 |
| K | Tumor carrying capacity | mm3 | 1423.1 |
| a | CTLs’ killing rate | mm3/(cell·day) | 0.0177 |
| d | Clearance rate of dying cells | 1/day | 0.264 |
| Fraction of viable cancer cells that survive | |||
| after radiation dose | |||
| Gy | - | 0.265 | |
| Gy | - | 0.664 | |
| Gy | - | 0.783 | |
| Fraction of cells that will undergo | |||
| immunogenic cell death after | |||
| radiation dose | |||
| Gy | - | 0.194 | |
| Gy | - | 0.984 | |
| Gy | - | 0.367 | |
| l | Decay rate of effector cells | 1/day | 0.03 |
| w | Baseline T cell recruitment rate | cell/ (mm3·day) | 0.135 |
| Fold change in the baseline T cell recruitment | |||
| rate due to immunogenic cell death | - | 15.37 | |
| e | Initial fold change in recruitment | ||
| of cytotoxic T cells caused by the administered | |||
| dose of 9H10 immunotherapy | - | 8.495 | |
| 9H10 immunotherapy clearance rate | 1/day | 0.967 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poleszczuk, J.; Enderling, H. The Optimal Radiation Dose to Induce Robust Systemic Anti-Tumor Immunity. Int. J. Mol. Sci. 2018, 19, 3377. https://doi.org/10.3390/ijms19113377
Poleszczuk J, Enderling H. The Optimal Radiation Dose to Induce Robust Systemic Anti-Tumor Immunity. International Journal of Molecular Sciences. 2018; 19(11):3377. https://doi.org/10.3390/ijms19113377
Chicago/Turabian StylePoleszczuk, Jan, and Heiko Enderling. 2018. "The Optimal Radiation Dose to Induce Robust Systemic Anti-Tumor Immunity" International Journal of Molecular Sciences 19, no. 11: 3377. https://doi.org/10.3390/ijms19113377
APA StylePoleszczuk, J., & Enderling, H. (2018). The Optimal Radiation Dose to Induce Robust Systemic Anti-Tumor Immunity. International Journal of Molecular Sciences, 19(11), 3377. https://doi.org/10.3390/ijms19113377