PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation


Abstract
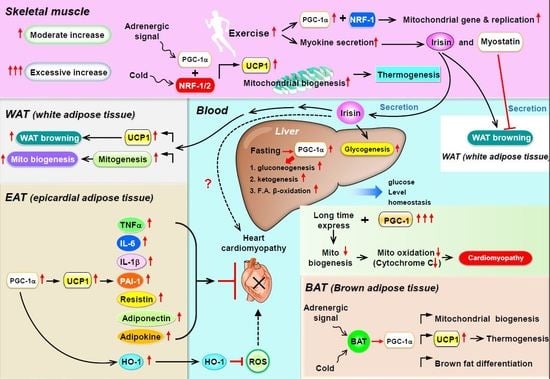
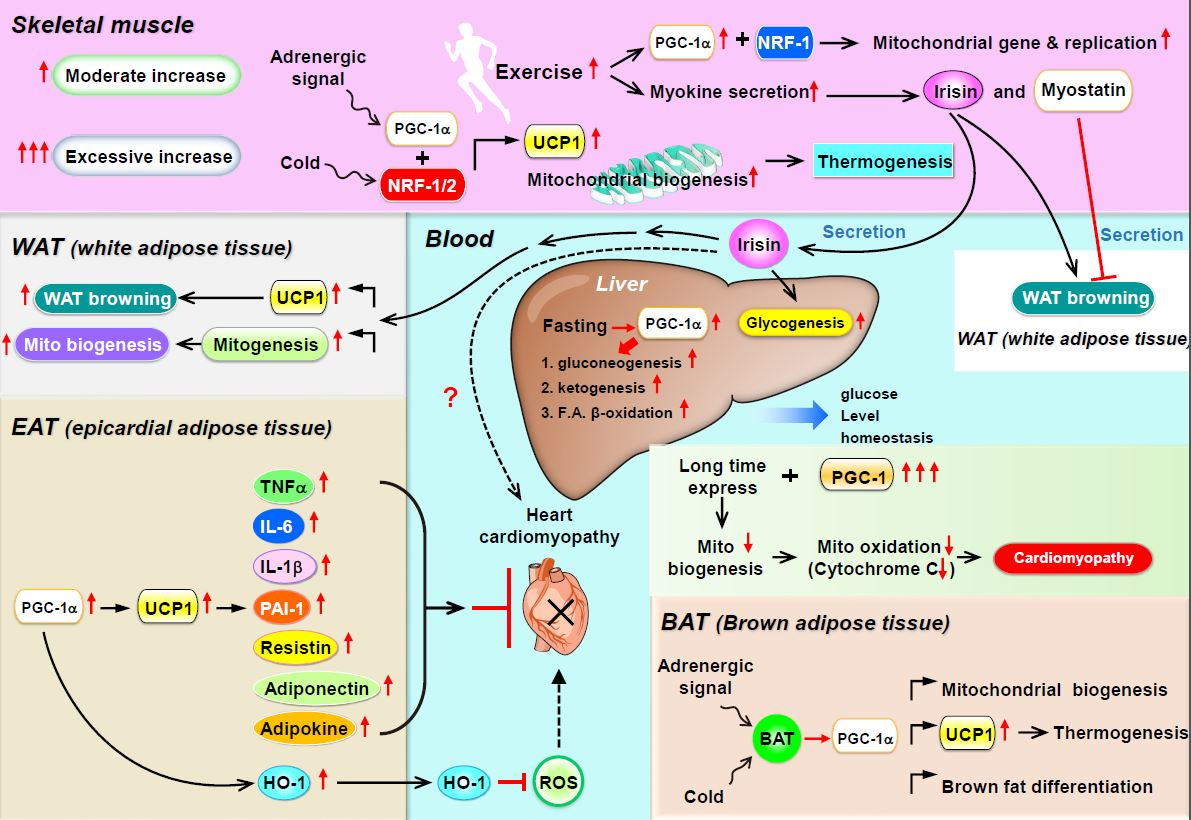
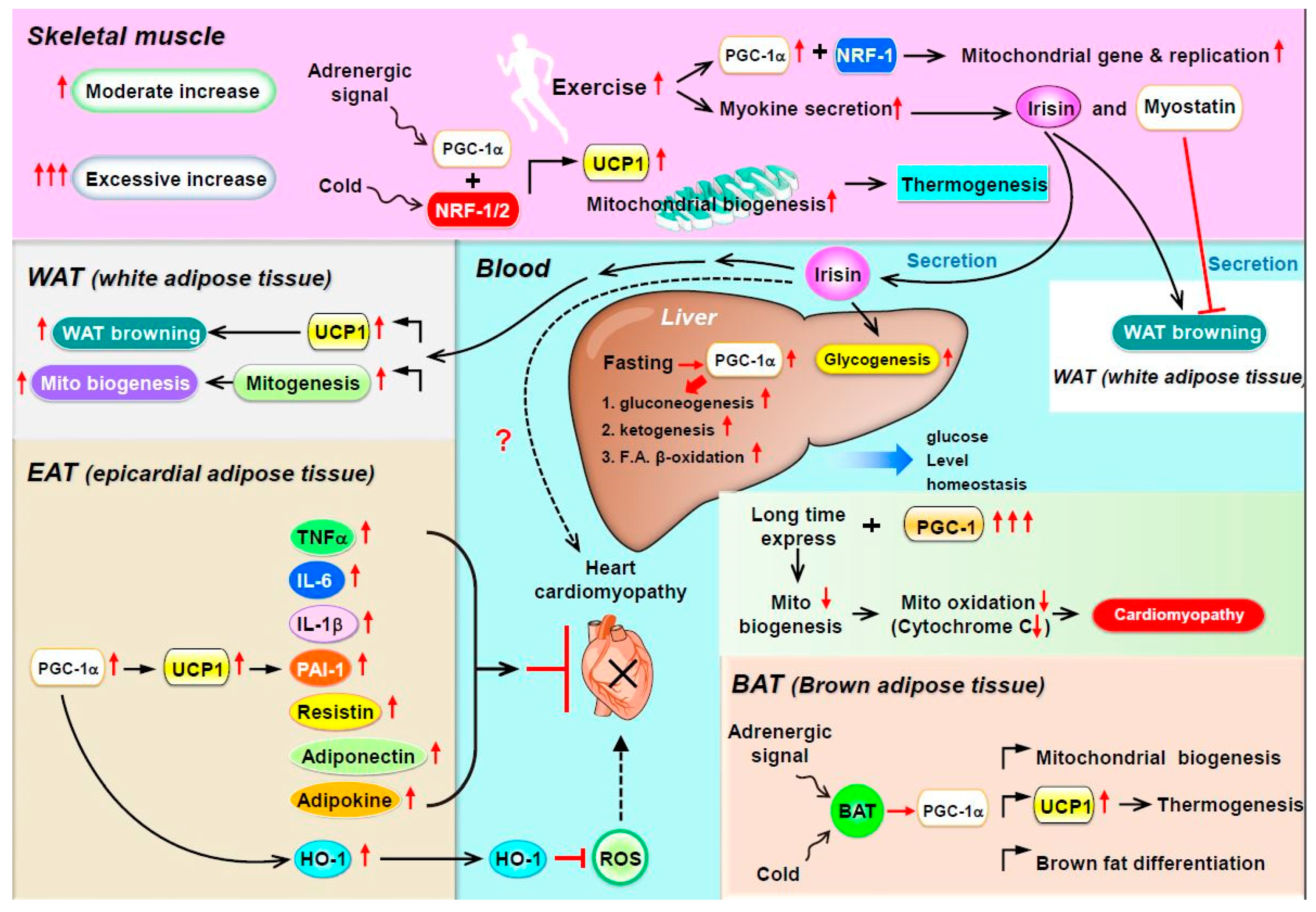
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. PGC-1α Can Regulate Lipid Metabolism
3. PGC-1α as a Coactivator for Metabolic Homeostasis in Skeletal Muscle
4. PGC-1α as a Coactivator in WAT Browning, Thermogenesis, and Mitochondrial Biogenesis
5. PGC-1 Controls Cardiac Energy Metabolism in Healthy or Diseased Myocardia
6. Is PGC-1 a Paracrine Regulator in Epicardial Adipose Tissue?
7. Potential for PGC-1α to Modulate Paracrine Regulators in the Heart
8. PGC-1α Regulates Metabolic Homeostasis in the Liver
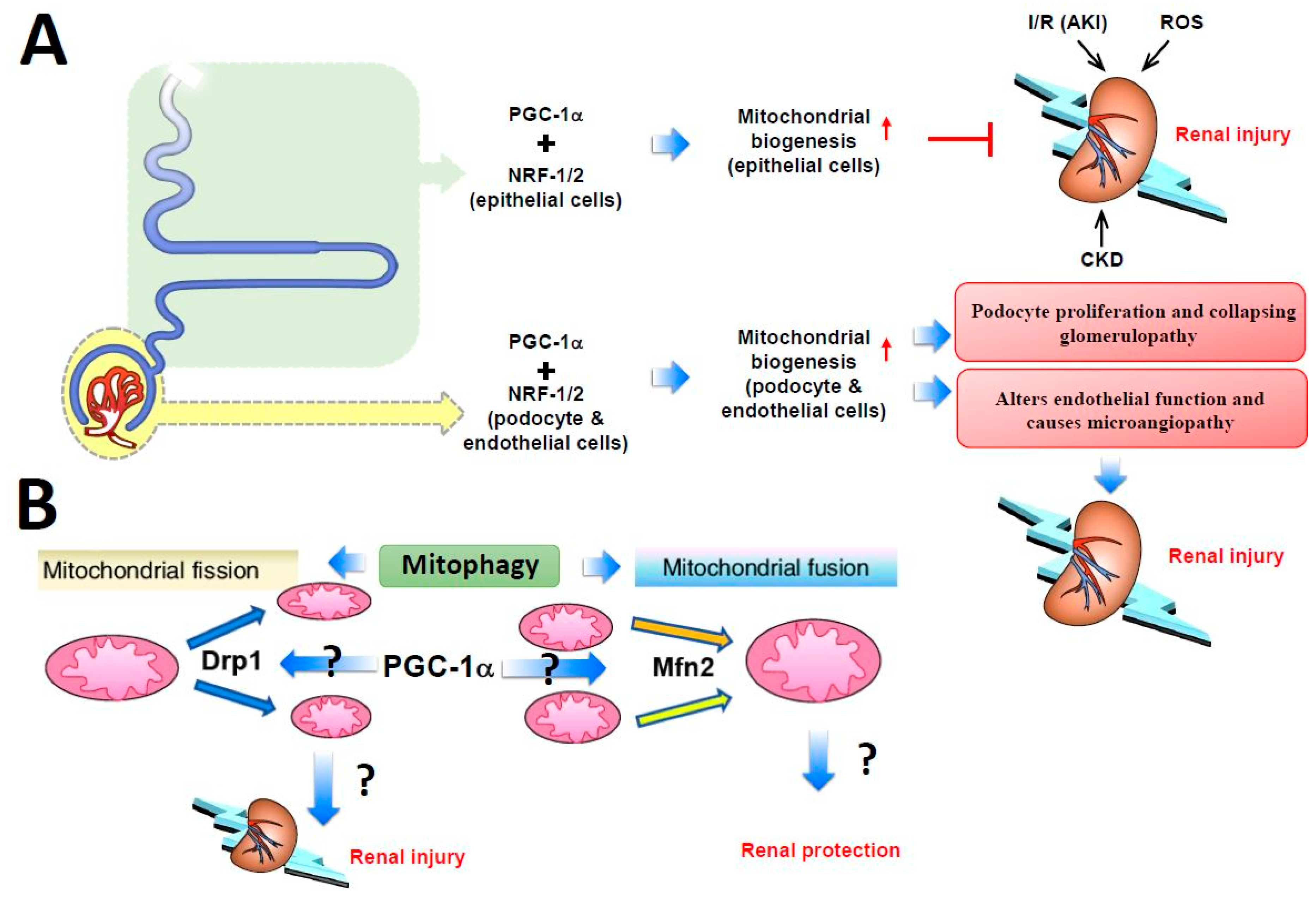
9. PGC-1α Regulates Kidney Metabolism via Mitochondrial Homeostasis
10. PGC-1α Regulates Cancer Metabolism
11. Conclusions
Funding
Conflicts of Interest
References
- Besseiche, A.; Riveline, J.P.; Gautier, J.F.; Breant, B.; Blondeau, B. Metabolic roles of PGC-1alpha and its implications for type 2 diabetes. Diabetes Metab. 2015, 41, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Popov, D.V.; Lysenko, E.A.; Kuzmin, I.V.; Vinogradova, V.; Grigoriev, A.I. Regulation of PGC-1alpha Isoform Expression in Skeletal Muscles. Acta Nat. 2015, 7, 48–59. [Google Scholar]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatazawa, Y.; Senoo, N.; Tadaishi, M.; Ogawa, Y.; Ezaki, O.; Kamei, Y.; Miura, S. Metabolomic Analysis of the Skeletal Muscle of Mice Overexpressing PGC-1alpha. PLoS ONE 2015, 10, e0129084. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.A.; Daniels, T.G.; Wang, X.; Paul, A.; Lin, J.; Spiegelman, B.M.; Stevenson, S.C.; Rangwala, S.M. Muscle-specific expression of PPARgamma coactivator-1alpha improves exercise performance and increases peak oxygen uptake. J. Appl. Physiol. 2008, 104, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.M.; Rebelo, A.P.; Moraes, C.T. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 2012, 64, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.S.; Befroy, D.E.; Codella, R.; Kim, S.; Reznick, R.M.; Hwang, Y.J.; Liu, Z.X.; Lee, H.Y.; Distefano, A.; Samuel, V.T.; et al. Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. USA 2008, 105, 19926–19931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza, D.O.; Boros, L.G.; Crunkhorn, S.; Gami, H.; Patti, M.E. Dual modulation of both lipid oxidation and synthesis by peroxisome proliferator-activated receptor-gamma coactivator-1alpha and -1beta in cultured myotubes. FASEB J. 2010, 24, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Mukai, K.; Lally, J.S.; Maher, A.C.; Gurd, B.J.; Heigenhauser, G.J.; Spriet, L.L.; Holloway, G.P. AMP-activated protein kinase is required for exercise-induced peroxisome proliferator-activated receptor co-activator 1 translocation to subsarcolemmal mitochondria in skeletal muscle. J. Physiol. 2013, 591, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Zheng, D.; Houmard, J.A.; Brault, J.J.; Hickner, R.C.; Cortright, R.N. Overexpression of PGC-1alpha increases peroxisomal activity and mitochondrial fatty acid oxidation in human primary myotubes. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E253–E263. [Google Scholar] [CrossRef] [PubMed]
- Summermatter, S.; Baum, O.; Santos, G.; Hoppeler, H.; Handschin, C. Peroxisome proliferator-activated receptor {gamma} coactivator 1{alpha} (PGC-1{alpha}) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J. Boil. Chem. 2010, 285, 32793–32800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supruniuk, E.; Miklosz, A.; Chabowski, A. The Implication of PGC-1alpha on Fatty Acid Transport across Plasma and Mitochondrial Membranes in the Insulin Sensitive Tissues. Front. Physiol. 2017, 8, 923. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Thomason, D.B. Molecular and cellular adaptation of muscle in response to exercise: Perspectives of various models. Physiol. Rev. 1991, 71, 541–585. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A. Invited Review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. 2001, 90, 1137–1157. [Google Scholar] [CrossRef] [PubMed]
- Bassel-Duby, R.; Olson, E.N. Signaling pathways in skeletal muscle remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef] [PubMed]
- Thom, R.; Rowe, G.C.; Jang, C.; Safdar, A.; Arany, Z. Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor gamma coactivator (PGC)-1alpha. J. Boil. Chem. 2014, 289, 8810–8817. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Chin, S.; Li, P.; Liu, F.; Maratos-Flier, E.; Lebrasseur, N.K.; Yan, Z.; Spiegelman, B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J. Boil. Chem. 2007, 282, 30014–30021. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Gupta, R.K.; Ruas, J.L.; Wu, J.; Naseri, E.; Estall, J.L.; Spiegelman, B.M. PGC-1alpha regulates a HIF2alpha-dependent switch in skeletal muscle fiber types. Proc. Natl. Acad. Sci. USA 2010, 107, 21866–21871. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.Y. The role of exercise-induced myokines in regulating metabolism. Arch. Pharm. Res. 2018, 41, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daskalopoulou, S.S.; Cooke, A.B.; Gomez, Y.H.; Mutter, A.F.; Filippaios, A.; Mesfum, E.T.; Mantzoros, C.S. Plasma irisin levels progressively increase in response to increasing exercise workloads in young, healthy, active subjects. Eur. J. Endocrinol. 2014, 171, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedrychowski, M.P.; Wrann, C.D.; Paulo, J.A.; Gerber, K.K.; Szpyt, J.; Robinson, M.M.; Nair, K.S.; Gygi, S.P.; Spiegelman, B.M. Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab. 2015, 22, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.Y.; Mougios, V.; Kabasakalis, A.; Fatouros, I.; Siopi, A.; Douroudos, I.I.; Filippaios, A.; Panagiotou, G.; Park, K.H.; Mantzoros, C.S. Exercise-induced irisin secretion is independent of age or fitness level and increased irisin may directly modulate muscle metabolism through AMPK activation. J. Clin. Endocrinol. Metab. 2014, 99, E2154–E2161. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Linderman, J.D.; Smith, S.; Brychta, R.J.; Wang, J.; Idelson, C.; Perron, R.M.; Werner, C.D.; Phan, G.Q.; Kammula, U.S.; et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 2014, 19, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Becerril, S.; Mendez-Gimenez, L.; Ramirez, B.; Sainz, N.; Catalan, V.; Gomez-Ambrosi, J.; Fruhbeck, G. Leptin administration activates irisin-induced myogenesis via nitric oxide-dependent mechanisms, but reduces its effect on subcutaneous fat browning in mice. Int. J. Obes. 2015, 39, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Dincer, F.; Mesfum, E.; Mantzoros, C.S. Irisin stimulates muscle growth-related genes and regulates adipocyte differentiation and metabolism in humans. Int. J. Obes. 2014, 38, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Shi, C.X.; Gao, R.; Sun, H.J.; Xiong, X.Q.; Ding, L.; Chen, Q.; Li, Y.H.; Wang, J.J.; Kang, Y.M.; et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin. Sci. 2015, 129, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhang, R.; Jiang, F.; Wang, J.; Chen, M.; Peng, D.; Yan, J.; Wang, S.; Bao, Y.; Hu, C.; et al. Circulating irisin levels are associated with lipid and uric acid metabolism in a Chinese population. Clin. Exp. Pharmacol. Physiol. 2015, 42, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Liu, J.; Zhang, J.; Zhu, D.; Wang, H.; Xiong, L.; Lee, Y.; Ye, J.; Lian, K.; Xu, C.; et al. Irisin improves fatty acid oxidation and glucose utilization in type 2 diabetes by regulating the AMPK signaling pathway. Int. J. Obes. 2016, 40, 443–451. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.L.; Hittel, D.S.; McPherron, A.C. Expression and function of myostatin in obesity, diabetes, and exercise adaptation. Med. Sci. Sports Exerc. 2011, 43, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Streeper, R.S.; Farese, R.V., Jr.; Yamamoto, K.R. Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects. Proc. Natl. Acad. Sci. USA 2006, 103, 15675–15680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wall, R.J.; Yang, J. Transgenic expression of myostatin propeptide prevents diet-induced obesity and insulin resistance. Biochem. Biophys. Res. Commun. 2005, 337, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; McFarlane, C.; Lokireddy, S.; Masuda, S.; Ge, X.; Gluckman, P.D.; Sharma, M.; Kambadur, R. Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia 2012, 55, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Dong, Y.; Dong, Y.; Chen, F.; Mitch, W.E.; Zhang, L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int. J. Obes. 2016, 40, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Liang, X.; Bi, P.; Kuang, S. Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1alpha-Fndc5 pathway in muscle. FASEB J. 2013, 27, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A.; Hefti, F. BDNF and NGF treatment in lesioned rats: Effects on cholinergic function and weight gain. Neuroreport 1992, 3, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Choi, E.Y.; Liu, X.; Martin, A.; Wang, C.; Xu, X.; During, M.J. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab. 2011, 14, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Jacobsson, A.; Rehnmark, S.; Nedergaard, J. Signal transduction in brown adipose tissue recruitment: Noradrenaline and beyond. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1996, 20 (Suppl. 3), S36–S42. [Google Scholar]
- Ricquier, D. Molecular biology of brown adipose tissue. Proc. Nutr. Soc. 1989, 48, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Boil. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Park, P.H.; Huang, H.; McMullen, M.R.; Mandal, P.; Sun, L.; Nagy, L.E. Suppression of lipopolysaccharide-stimulated tumor necrosis factor-alpha production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. J. Boil. Chem. 2008, 283, 26850–26858. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.S.; Dudycha, S.J.; O’Rourke, B. Cardiac energy metabolism: Models of cellular respiration. Annu. Rev. Biomed. Eng. 2001, 3, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Energy metabolism in heart failure. J. Physiol. 2004, 555 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic gene expression in fetal and failing human heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Fortin, D.; Delomenie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003, 551 Pt 2, 491–501. [Google Scholar] [CrossRef]
- Goffart, S.; von Kleist-Retzow, J.C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguere, V.; Kelly, D.P. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Boil. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Nuclear control of respiratory gene expression in mammalian cells. J. Cell. Biochem. 2006, 97, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Tuomainen, T.; Tavi, P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp. Cell Res. 2017, 360, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 2010, 107, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Boil. 2000, 20, 1868–1876. [Google Scholar] [CrossRef]
- Duncan, J.G.; Fong, J.L.; Medeiros, D.M.; Finck, B.N.; Kelly, D.P. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation 2007, 115, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Boil. 2005, 3, e101. [Google Scholar]
- Russell, L.K.; Mansfield, C.M.; Lehman, J.J.; Kovacs, A.; Courtois, M.; Saffitz, J.E.; Medeiros, D.M.; Valencik, M.L.; McDonald, J.A.; Kelly, D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004, 94, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267 Pt 2, H742–H750. [Google Scholar] [CrossRef]
- Christe, M.E.; Rodgers, R.L. Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J. Mol. Cell. Cardiol. 1994, 26, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; Overturf, M.L. Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension 1988, 11, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Massie, B.M.; Schaefer, S.; Garcia, J.; McKirnan, M.D.; Schwartz, G.G.; Wisneski, J.A.; Weiner, M.W.; White, F.C. Myocardial high-energy phosphate and substrate metabolism in swine with moderate left ventricular hypertrophy. Circulation 1995, 91, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Razeghi, P.; Essop, M.F.; Huss, J.M.; Abbasi, S.; Manga, N.; Taegtmeyer, H. Hypoxia-induced switches of myosin heavy chain iso-gene expression in rat heart. Biochem. Biophys. Res. Commun. 2003, 303, 1024–1027. [Google Scholar] [CrossRef]
- Van Bilsen, M.; Smeets, P.J.; Gilde, A.J.; van der Vusse, G.J. Metabolic remodelling of the failing heart: The cardiac burn-out syndrome? Cardiovasc. Res. 2004, 61, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Van Bilsen, M. “Energenetics” of heart failure. Ann. N. Y. Acad. Sci. 2004, 1015, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E.; Laws, F.A.; Goodwin, G.W.; Taegtmeyer, H. Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J. Boil. Chem. 2001, 276, 44390–44395. [Google Scholar] [CrossRef] [PubMed]
- Remondino, A.; Rosenblatt-Velin, N.; Montessuit, C.; Tardy, I.; Papageorgiou, I.; Dorsaz, P.A.; Jorge-Costa, M.; Lerch, R. Altered expression of proteins of metabolic regulation during remodeling of the left ventricle after myocardial infarction. J. Mol. Cell. Cardiol. 2000, 32, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt-Velin, N.; Montessuit, C.; Papageorgiou, I.; Terrand, J.; Lerch, R. Postinfarction heart failure in rats is associated with upregulation of GLUT-1 and downregulation of genes of fatty acid metabolism. Cardiovasc. Res. 2001, 52, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Dewald, O.; Sharma, S.; Adrogue, J.; Salazar, R.; Duerr, G.D.; Crapo, J.D.; Entman, M.L.; Taegtmeyer, H. Downregulation of peroxisome proliferator-activated receptor-alpha gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation 2005, 112, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Wang, S.C.; Shirai, M.; Scaglia, F.; Xie, M.; Sakai, S.; Tanaka, T.; Kulkarni, P.A.; Barger, P.M.; Youker, K.A.; et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 2004, 23, 3559–3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiguchi, K.; Tian, Q.; Ishiyama, M.; Burchfield, J.; Gao, F.; Mann, D.L.; Barger, P.M. Inhibition of PPAR-alpha activity in mice with cardiac-restricted expression of tumor necrosis factor: Potential role of TGF-beta/Smad3. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1443–H1451. [Google Scholar] [CrossRef] [PubMed]
- Pellieux, C.; Aasum, E.; Larsen, T.S.; Montessuit, C.; Papageorgiou, I.; Pedrazzini, T.; Lerch, R. Overexpression of angiotensinogen in the myocardium induces downregulation of the fatty acid oxidation pathway. J. Mol. Cell. Cardiol. 2006, 41, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Levy, F.H.; Kelly, D.P. Hypoxia inhibits the peroxisome proliferator-activated receptor alpha/retinoid X receptor gene regulatory pathway in cardiac myocytes: A mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. J. Boil. Chem. 2001, 276, 27605–27612. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R.S.; Hill, J.A. The use of ranolazine in cardiovascular disease. Expert Opin. Investig. Drugs 2002, 11, 117–123. [Google Scholar] [PubMed]
- Rupp, H.; Zarain-Herzberg, A.; Maisch, B. The use of partial fatty acid oxidation inhibitors for metabolic therapy of angina pectoris and heart failure. Herz 2002, 27, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.P.; Stanley, W.C.; Morita, H.; Suzuki, G.; Roth, B.A.; Blackburn, B.; Wolff, A.; Sabbah, H.N. Short-term treatment with ranolazine improves mechanical efficiency in dogs with chronic heart failure. Circ. Res. 2002, 91, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Jain, M.; Cui, L.; D’Agostino, J.; Aiello, F.; Luptak, I.; Ngoy, S.; Mortensen, R.M.; Tian, R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002, 106, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Takano, H.; Nagai, T.; Uozumi, H.; Hasegawa, H.; Kubota, N.; Saito, T.; Masuda, Y.; Kadowaki, T.; Komuro, I. Peroxisome proliferator-activated receptor gamma plays a critical role in inhibition of cardiac hypertrophy in vitro and in vivo. Circulation 2002, 105, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ohki, R.; Lee, R.T.; Ikeda, U.; Shimada, K. Peroxisome proliferator-activated receptor gamma activators inhibit cardiac hypertrophy in cardiac myocytes. Circulation 2001, 104, 1670–1675. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Rodriguez-Calvo, R.; Jove, M.; Michalik, L.; Wahli, W.; Laguna, J.C.; Vazquez-Carrera, M. Peroxisome proliferator-activated receptor beta/delta activation inhibits hypertrophy in neonatal rat cardiomyocytes. Cardiovasc. Res. 2005, 65, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Fein, F.S.; Sonnenblick, E.H. Diabetic cardiomyopathy. Cardiovasc. Drugs Ther. 1994, 8, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [PubMed]
- Keen, H.; Jarrett, R.J. The WHO multinational study of vascular disease in diabetes: 2. Macrovascular disease prevalence. Diabetes Care 1979, 2, 187–195. [Google Scholar] [PubMed]
- Fein, F.S.; Sonnenblick, E.H. Diabetic cardiomyopathy. Prog. Cardiovasc. Dis. 1985, 27, 255–270. [Google Scholar] [CrossRef]
- Gamble, J.; Lopaschuk, G.D. Glycolysis and glucose oxidation during reperfusion of ischemic hearts from diabetic rats. Biochim. Biophys. Acta 1994, 1225, 191–199. [Google Scholar] [CrossRef]
- Stanley, W.C.; Lopaschuk, G.D.; McCormack, J.G. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc. Res. 1997, 34, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Belke, D.D.; Larsen, T.S.; Gibbs, E.M.; Severson, D.L. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1104–E1113. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, B.; McNeill, J.H. The diabetic heart: Metabolic causes for the development of a cardiomyopathy. Cardiovasc. Res. 1992, 26, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, C.; Weng, S.; Feng, C.; Finck, B.N.; Knutsen, R.H.; Leone, T.C.; Coleman, T.; Mecham, R.P.; Kelly, D.P.; Semenkovich, C.F. Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat. Med. 2003, 9, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.H.; Moore, K.H.; Giacomelli, F.; Wiener, J. Defective oxidative metabolism of heart mitochondria from genetically diabetic mice. Diabetes 1983, 32, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Konno, N.; Kako, K.J. Mitochondrial dysfunction observed in situ in cardiomyocytes of rats in experimental diabetes. Cardiovasc. Res. 1992, 26, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; O’Neill, B.T.; Tathireddy, P.; Young, M.E.; Abel, E.D. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005, 112, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G. Epicardial and pericardial fat: Close, but very different. Obesity 2009, 17, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, T.P.; Czech, M.P. Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: Basic mechanisms and clinical associations. J. Am. Heart Assoc. 2014, 3, e000582. [Google Scholar] [CrossRef] [PubMed]
- Sacks, H.S.; Fain, J.N.; Holman, B.; Cheema, P.; Chary, A.; Parks, F.; Karas, J.; Optican, R.; Bahouth, S.W.; Garrett, E.; et al. Uncoupling protein-1 and related messenger ribonucleic acids in human epicardial and other adipose tissues: Epicardial fat functioning as brown fat. J. Clin. Endocrinol. Metab. 2009, 94, 3611–3615. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Mori, J.; McLean, B.A.; Basu, R.; Das, S.K.; Ramprasath, T.; Parajuli, N.; Penninger, J.M.; Grant, M.B.; Lopaschuk, G.D.; et al. ACE2 Deficiency Worsens Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Response to Diet-Induced Obesity. Diabetes 2016, 65, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Lopaschuk, G.D.; Carvalho, E. Cellular cross-talk between epicardial adipose tissue and myocardium in relation to the pathogenesis of cardiovascular disease. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E937–E949. [Google Scholar] [CrossRef] [PubMed]
- Nour-Eldine, W.; Ghantous, C.M.; Zibara, K.; Dib, L.; Issaa, H.; Itani, H.A.; El-Zein, N.; Zeidan, A. Adiponectin Attenuates Angiotensin II-Induced Vascular Smooth Muscle Cell Remodeling through Nitric Oxide and the RhoA/ROCK Pathway. Front. Pharmacol. 2016, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.; Ahswin, H.; Smart, N.; Bayon, Y.; Wohlert, S.; Hunt, J.A. Reactive oxygen species (ROS)—A family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur. Cells Mater. 2012, 24, 249–265. [Google Scholar] [CrossRef]
- Iacobellis, G.; Pistilli, D.; Gucciardo, M.; Leonetti, F.; Miraldi, F.; Brancaccio, G.; Gallo, P.; di Gioia, C.R. Adiponectin expression in human epicardial adipose tissue in vivo is lower in patients with coronary artery disease. Cytokine 2005, 29, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Silaghi, A.; Achard, V.; Paulmyer-Lacroix, O.; Scridon, T.; Tassistro, V.; Duncea, I.; Clement, K.; Dutour, A.; Grino, M. Expression of adrenomedullin in human epicardial adipose tissue: Role of coronary status. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1443–E1450. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Bianco, A.C. Epicardial adipose tissue: Emerging physiological, pathophysiological and clinical features. Trends Endocrinol. Metab. TEM 2011, 22, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; di Gioia, C.R.; Cotesta, D.; Petramala, L.; Travaglini, C.; De Santis, V.; Vitale, D.; Tritapepe, L.; Letizia, C. Epicardial adipose tissue adiponectin expression is related to intracoronary adiponectin levels. Horm. Metab. Res. 2009, 41, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lian, W.S.; Chen, H.H.; Lai, P.F.; Cheng, C.F. Adiponectin ameliorates iron-overload cardiomyopathy through the PPARalpha-PGC-1-dependent signaling pathway. Mol. Pharmacol. 2013, 84, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Kroller-Schon, S.; Li, C.; Kant, S.; Cai, S.; Chen, K.; Contractor, M.M.; Pei, Y.; Schulz, E.; Keaney, J.F., Jr. PGC-1alpha dictates endothelial function through regulation of eNOS expression. Sci. Rep. 2016, 6, 38210. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Horton, J.L.; Kelly, D.P. Maintaining ancient organelles: Mitochondrial biogenesis and maturation. Circ. Res. 2015, 116, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Kelly, D.P. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin. Exp. Pharmacol. Physiol. 2002, 29, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [PubMed]
- Portilla, D.; Dai, G.; McClure, T.; Bates, L.; Kurten, R.; Megyesi, J.; Price, P.; Li, S. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 2002, 62, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Noireaud, J.; Andriantsitohaina, R. Recent insights in the paracrine modulation of cardiomyocyte contractility by cardiac endothelial cells. BioMed Res. Int. 2014, 2014, 923805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Pich, S.; Paz, J.C.; Sanches, R.; Martinez, V.; Orpinell, M.; Palacin, M.; Zorzano, A.; Guma, A. Neuregulins increase mitochondrial oxidative capacity and insulin sensitivity in skeletal muscle cells. Diabetes 2007, 56, 2185–2193. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Tarr, P.T.; Yang, R.; Rhee, J.; Puigserver, P.; Newgard, C.B.; Spiegelman, B.M. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J. Boil. Chem. 2003, 278, 30843–30848. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [PubMed]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.; Inoue, Y.; Yoon, J.C.; Puigserver, P.; Fan, M.; Gonzalez, F.J.; Spiegelman, B.M. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): Requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 4012–4017. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jager, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Satoh, H.; Herzig, S.; Lee, C.H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Arning, E.; Liu, C.; Vitvitsky, V.; Hernandez, C.; Banerjee, R.; Bottiglieri, T.; Lin, J.D. Regulation of homocysteine homeostasis through the transcriptional coactivator PGC-1alpha. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E543–E548. [Google Scholar] [CrossRef] [PubMed]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARgamma-coactivator 1alpha (PGC1alpha) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Haase, T.N.; Ringholm, S.; Leick, L.; Bienso, R.S.; Kiilerich, K.; Johansen, S.; Nielsen, M.M.; Wojtaszewski, J.F.; Hidalgo, J.; Pedersen, P.A.; et al. Role of PGC-1alpha in exercise and fasting-induced adaptations in mouse liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1501–R1509. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.A.; Eagon, J.C.; LaRiviere, L.L.; Korenblat, K.M.; Klein, S.; Finck, B.N. Hepatic lipin 1beta expression is diminished in insulin-resistant obese subjects and is reactivated by marked weight loss. Diabetes 2007, 56, 2395–2399. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.M.; Meers, G.M.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. PGC-1alpha overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G979–G992. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, S.; Liu, T.; Borjigin, J.; Lin, J.D. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature 2007, 447, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M. Mitochondrial biogenesis in kidney disease. J. Am. Soc. Nephrol. JASN 2011, 22, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Schnellmann, R.G. PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem. Biophys. Res. Commun. 2007, 355, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, J.; Zhou, F.; Wang, W.; Chen, N. PGC-1alpha ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol. Med. Rep. 2018, 17, 4490–4498. [Google Scholar] [PubMed]
- Sogabe, K.; Roeser, N.F.; Venkatachalam, M.A.; Weinberg, J.M. Differential cytoprotection by glycine against oxidant damage to proximal tubule cells. Kidney Int. 1996, 50, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Schnellmann, R.G. Signaling of mitochondrial biogenesis following oxidant injury. J. Boil. Chem. 2007, 282, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.I.; Kim, H.J.; Park, J.S.; Kim, I.J.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1alpha attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3beta inactivation mediated by activated p38 in HK-2 Cells. Sci. Rep. 2017, 7, 4319. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.; Cho, S.G.; Wang, C.Y.; Yang, T.; Dong, Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am. J. Physiol. Cell Physiol. 2011, 300, C447–C455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.H.; Guo, X.; Ma, D.; Guo, Y.; Li, Q.; Yang, D.; Li, P.; Qiu, X.; Wen, S.; Xiao, R.P.; et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Boil. 2004, 6, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Dasgupta, A.; Ding, J.; Indig, F.E.; Ghosh, P.; Longo, D.L. Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. FASEB J. 2014, 28, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chen, J.K.; Harris, R.C. Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int. 2012, 82, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Han, S.J.; Kim, J.I.; Lee, S.; Lipschutz, J.H.; Park, K.M. Activation of ERK accelerates repair of renal tubular epithelial cells, whereas it inhibits progression of fibrosis following ischemia/reperfusion injury. Biochim. Biophys. Acta 2013, 1832, 1998–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, J.M.; Wang, Z.; Bonegio, R.G.; Havasi, A.; Liesa, M.; Vemula, P.; Borkan, S.C. Conditional knockout of proximal tubule mitofusin 2 accelerates recovery and improves survival after renal ischemia. J. Am. Soc. Nephrol. JASN 2015, 26, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Boil. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. TIG 2017, 33, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 2007, 447, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.; Gravel, S.P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.D.; Jones, R.G.; et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Watkins, G.; Douglas-Jones, A.; Mansel, R.E.; Jiang, W.G. The localisation and reduction of nuclear staining of PPARgamma and PGC-1 in human breast cancer. Oncol. Rep. 2004, 12, 483–488. [Google Scholar] [PubMed]
- Jiang, W.G.; Douglas-Jones, A.; Mansel, R.E. Expression of peroxisome-proliferator activated receptor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. Int. J. Cancer 2003, 106, 752–757. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Boil. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabaries, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chenard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1alpha Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Lim, J.H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, I.; Salvatore, L.; Murzilli, S.; Lo Sasso, G.; Latorre, D.; Martelli, N.; Egorova, A.V.; Polishuck, R.; Madeyski-Bengtson, K.; Lelliott, C.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc. Natl. Acad. Sci. USA 2011, 108, 6603–6608. [Google Scholar] [CrossRef] [PubMed]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrielson, M.; Bjorklund, M.; Carlson, J.; Shoshan, M. Expression of mitochondrial regulators PGC1alpha and TFAM as putative markers of subtype and chemoresistance in epithelial ovarian carcinoma. PLoS ONE 2014, 9, e107109. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Jung, J.W.; Jung, J.; Han, Y.; Suh, D.H.; Kim, H.S.; Dhanasekaran, D.N.; Song, Y.S. PGC1alpha induced by reactive oxygen species contributes to chemoresistance of ovarian cancer cells. Oncotarget 2017, 8, 60299–60311. [Google Scholar] [PubMed]
- Liu, R.; Zhang, H.; Zhang, Y.; Li, S.; Wang, X.; Wang, X.; Wang, C.; Liu, B.; Zen, K.; Zhang, C.Y.; et al. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha acts as a tumor suppressor in hepatocellular carcinoma. Tumour Boil. 2017, 39, 1010428317695031. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.F.; Xu, C.; Pan, X.; Cai, L.; Lin, X.Y.; Chen, S.; Biskup, E. Prognostic value of plasma levels of HIF-1a and PGC-1a in breast cancer. Oncotarget 2016, 7, 77793–77806. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene 2014, 33, 5251–5261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ba, Y.; Liu, C.; Sun, G.; Ding, L.; Gao, S.; Hao, J.; Yu, Z.; Zhang, J.; Zen, K.; et al. PGC-1alpha induces apoptosis in human epithelial ovarian cancer cells through a PPARgamma-dependent pathway. Cell Res. 2007, 17, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Moraes, C.T. Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels. Mol. Oncol. 2011, 5, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Su, Y.; Yin, P.H.; Lee, H.C.; Chi, C.W. PPAR(gamma)/PGC-1(alpha) pathway in E-cadherin expression and motility of HepG2 cells. Anticancer. Res. 2009, 29, 5057–5063. [Google Scholar] [PubMed]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernandez-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat. Cell Boil. 2016, 18, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Tada, Y.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Uchiumi, T.; Fujimoto, N.; Seki, N.; Naito, S. Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol. Endocrinol. 2010, 24, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.W.; Yun, S.H.; Park, E.S.; Jeong, J.S.; Kwak, J.Y.; Park, J.I. Overexpression of PGC1alpha enhances cell proliferation and tumorigenesis of HEK293 cells through the upregulation of Sp1 and Acyl-CoA binding protein. Int. J. Oncol. 2015, 46, 1328–1342. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Delgado, O.; Celiktas, M.; Katayama, H.; Wang, H.; Gazdar, A.F.; Hanash, S.M. Proteomic signatures associated with p53 mutational status in lung adenocarcinoma. Proteomics 2014, 14, 2750–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, C.-F.; Ku, H.-C.; Lin, H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. https://doi.org/10.3390/ijms19113447
Cheng C-F, Ku H-C, Lin H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. International Journal of Molecular Sciences. 2018; 19(11):3447. https://doi.org/10.3390/ijms19113447
Chicago/Turabian StyleCheng, Ching-Feng, Hui-Chen Ku, and Heng Lin. 2018. "PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation" International Journal of Molecular Sciences 19, no. 11: 3447. https://doi.org/10.3390/ijms19113447
APA StyleCheng, C. -F., Ku, H. -C., & Lin, H. (2018). PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. International Journal of Molecular Sciences, 19(11), 3447. https://doi.org/10.3390/ijms19113447