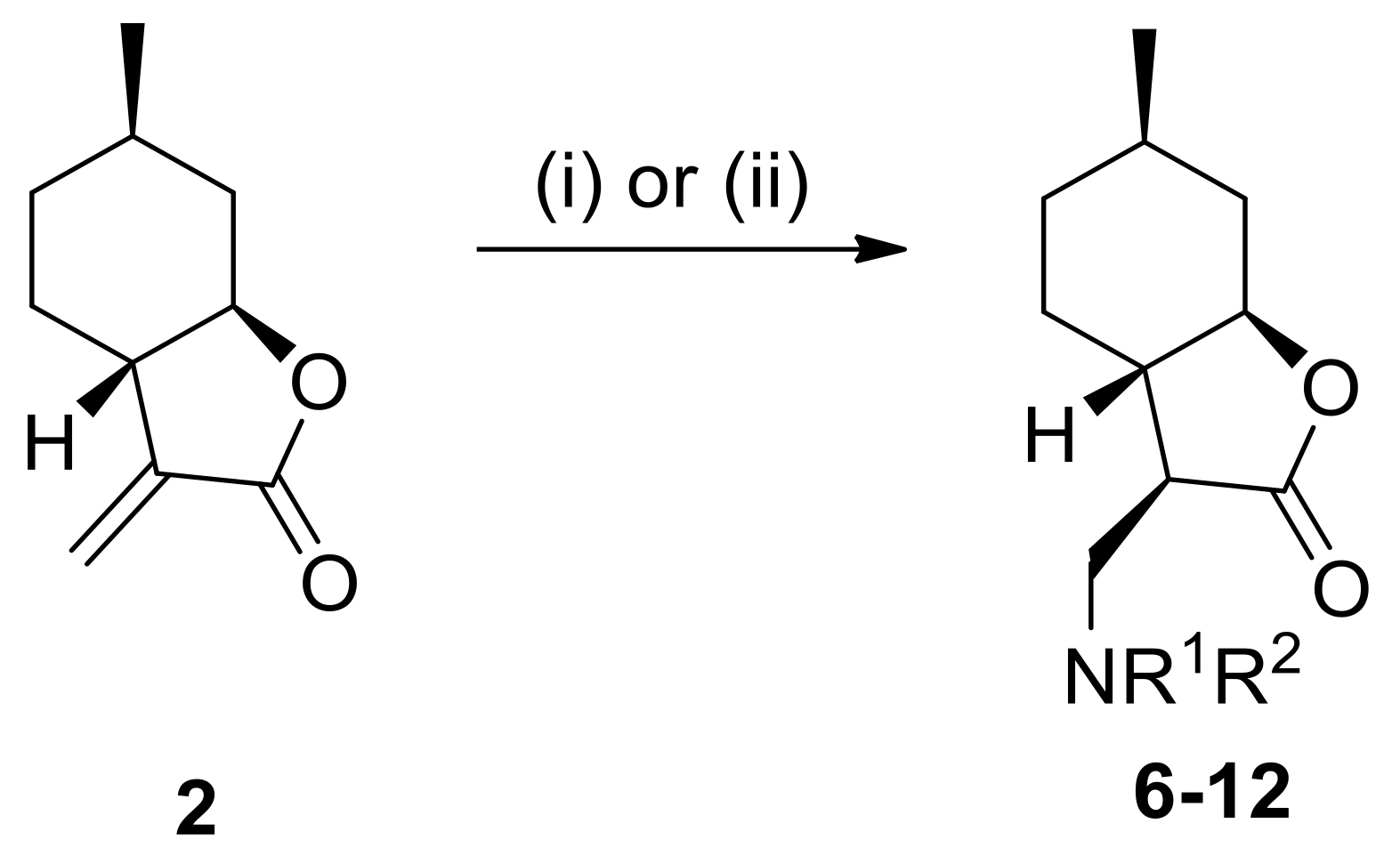
4.3. General Procedure for Nucleophilic Addition of α-Methylene-γ-Butyrolactone to Amines
Amines (1.2 mmol) were added to the solution of α-methylene-γ-butyrolactone 2 or 4 (1.2 mmol) in dry EtOH (2.0 mL). The reaction mixture was stirred at appropriate temperatures for 20–72 h. When the reaction was complete (indicated by TLC), EtOH was removed under reduced pressure. The crude residue was purified by column chromatography on silica gel with an appropriate solvent mixture. The crude products after solvent evaporation were purified as HCl salts by recrystallization in diethyl ether resulting in compounds 5a–10 and 13–18.
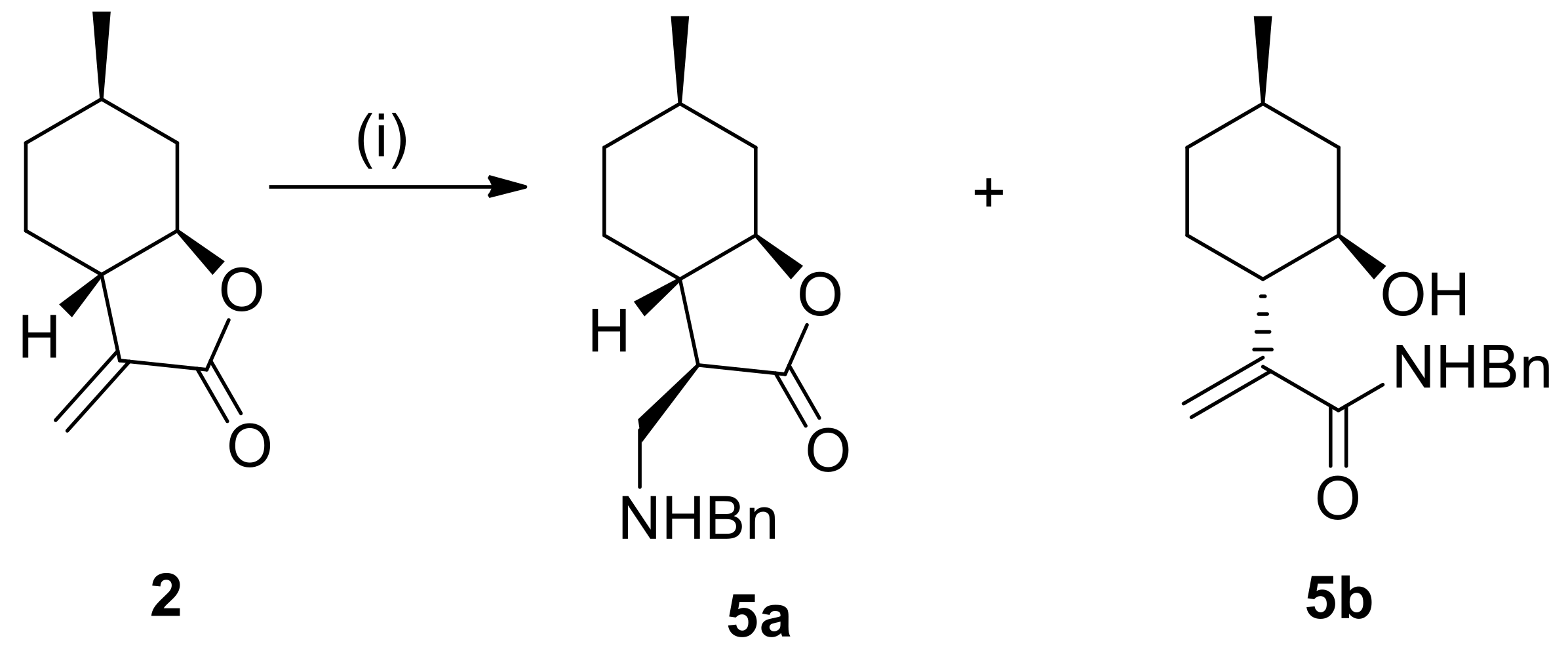
4.3.1. (3R,3aS,6R,7aR)-3-((Benzylamino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one hydrochloride (5a)
Prepared from 2 with benzylamine at 25 °C for 20 h. Compound 5a was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 65%, white crystals, m.p.: 190–198 °C. = −8.0 (c 0.23, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.94–1.00 (1H, m), 0.95 (3H, d, J = 8.1 Hz), 1.09–1.18 (1H, m), 1.24–1.32 (1H, m), 1.59–1.69 (2H, m), 1.78–1.86 (1H, m), 2.06–2.13 (2H, m), 3.05–3.19 (3H, m), 3.93 (1H, td, J = 4.4, 14.1 Hz), 7.41–7.63 (5H, m), 9.67 (2H, s). 13C NMR (125 MHz, DMSO-d6): δ = 21.9, 26.1, 30.5, 33.4, 37.6, 42.7, 44.6, 47.0, 50.5, 81.9, 128.5, 128.8, 130.2, 131.7, 176.1. Anal. Calcd for C17H24ClNO2: C, 65.90; H, 7.81; N, 4.52. Found: C, 65.85; H, 7.85; N, 4.52.
4.3.2. (3R,3aS,6R,7aR)-6-Methyl-3-((((R)-1-phenylethyl)amino)methyl)hexahydrobenzofuran-2(3H)-one hydrochloride (6)
Prepared from 2 with (R)-α-methylbenzylamine at 25 °C for 20 h. Compound 6 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 75%, white crystals, m.p.: 170–180 °C. = +21.0 (c 0.23, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.91–1.00 (1H, m), 0.95 (3H, d, J = 6.5 Hz), 1.08–1.15 (1H, m), 1.23–1.28 (1H, m), 1.53–1.62 (1H, m), 1.61 (3H, d, J = 6.4 Hz), 1.68 (1H, d, J = 13.2 Hz), 1.75–1.82 (1H, m), 2.03–2.12 (2H, m), 2.78–2.86 (1H, m), 2.94 (1H, t, J = 6.3 Hz), 3.05–3.15 (1H, m), 3.92 (1H, td, J = 3.4, 11.3 Hz), 4.38 (1H, s), 7.39–7.61 (5H, m), 9.17 (1H, br s), 9.83 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 19.4, 21.9, 26.0, 30.5, 33.4, 37.5, 46.4, 58.2, 81.9, 128.0, 128.8, 176.0. Anal. Calcd for C18H26ClNO2: C, 66.76; H, 8.09; N, 4.32. Found: C, 66.75; H, 8.04; N, 4.30.
4.3.3. (3R,3aS,6R,7aR)-6-Methyl-3-((((S)-1-phenylethyl)amino)methyl)hexahydrobenzofuran-2(3H)-one hydrochloride (7)
Prepared from 2 with (S)-α-methylbenzylamine at 25 °C for 20 h. Compound 7 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 71%, white crystals, m.p.: 170–180 °C. = −26.0 (c 0.24, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.91–0.97 (1H, m), 0.94 (1H, d, J = 6.6 Hz), 1.08–1.15 (1H, m), 1.19–1.28 (1H, m), 1.57–1.67 (2H, m), 1.62 (3H, d, J = 6.7 Hz), 1.74–1.81 (1H, m), 1.90–1.93 (1H, m), 2.09–2.11 (1H, m), 2.85–2.94 (1H, m), 2.95–3.02 (1H, m), 3.93 (1H, td, J = 3.5, 11.3 Hz), 4.36 (1H, br s), 7.39–7.64 (5H, m), 9.25 (1H, br s), 9.82 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 19.4, 21.9, 26.0, 30.5, 33.4, 37.5, 42.8, 43.5, 46.8, 58.3, 82.0, 127.9, 128.9, 137.1, 176.3. Anal. Calcd for C14H26ClNO2: C, 66.76; H, 8.09; N, 4.32. Found: C, 66.78; H, 8.10; N, 4.35.
4.3.4. (3R,3aS,6R,7aR)-3-((Diethylamino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one hydrochloride (8)
Prepared from 2 with diethylamine at 25 °C for 20 h. Compound 8 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 50%, colorless oil. = −7.0 (c 0.27, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.91–0.99 (1H, m), 0.96 (3H, d, J = 6.6 Hz), 1.05 (1H, t, J = 7.0 Hz), 1.14 (1H, q, J = 11.5 Hz), 1.24 (6H, td, J = 2.3, 7.1 Hz), 1.31–1.39 (1H, m), 1.58–1.63 (1H, m), 1.71–1.82 (2H, m), 1.97–2.01 (1H, m), 2.12–2.15 (1H, m), 3.12–3.23 (6H, m), 3.38–3.44 (2H, m), 3.97 (1H, td, J = 3.6, 11.4 Hz), 9.94 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 8.4, 8.5, 21.8, 25.6, 30.6, 33.4, 37.5, 41.3, 46.8, 47.2, 47.5, 49.1, 81.9, 176.8. Anal. Calcd for C14H26ClNO2: C, 60.96; H, 9.50; N, 5.08. Found: C, 60.70; H, 9.45; N, 5.10.
4.3.5. (3R,3aS,6R,7aR)-6-Methyl-3-(piperidin-1-ylmethyl)hexahydrobenzofuran-2(3H)-one hydrochloride (9)
Prepared from 2 with pyridine at 25 °C for 20 h. Compound 9 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 47%, white crystals, m.p.: 180–190 °C. = −3.3 (c 0.31, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.92–1.00 (1H, m), 0.96 (1H, d, J = 6.6 Hz), 1.14 (1H, q, J = 11.5 Hz), 1.30–1.40 (1H, m), 1.58–1.64 (1H, m), 1.67–1.83 (6H, m), 2.02–2.05 (1H, m), 2.11–2.13 (1H, m), 2.88–2.96 (2H, m), 3.16–3.21 (2H, m), 3.26–3.29 (1H, m), 3.42 (1H, d, J = 12.0 Hz), 3.94 (1H, td, J = 3.6, 11.3 Hz), 10.5 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 21.1, 21.8, 22.2, 25.8, 30.6, 33.4, 37.5, 41.4, 47.7, 51.7, 52.8, 53.8, 81.8, 176.7. Anal. Calcd for C15H26ClNO2: C, 62.59; H, 9.10; N, 4.87. Found: C, 62.60; H, 9.15; N, 4.90.
4.3.6. (3R,3aS,6R,7aR)-3-((Dibenzylamino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one hydrochloride (10)
Prepared from 2 with dibenzylamine at 70 °C for 72 h. Compound 10 was purified by column chromatography on silica gel (n-hexane/ethyl acetate = 9:1). Yield: 59%, white crystals, m.p.: 120–125 °C. = −27.0 (c 0.27, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.80–0.87 (1H, m), 0.93 (3H, d, J = 6.5 Hz), 1.10 (1H, q, J = 11.4 Hz), 1.23–1.31 (1H, m), 1.55–1.72 (4H, m), 2.09 (1H, d, J = 11.0 Hz), 3.09 (1H, d, J = 12.9 Hz), 3.33–3.40 (1H, m), 3.92 (1H, t, J = 9.6 Hz), 4.30–4.33 (2H, m), 4.44 (1H, d, J = 9.3 Hz), 7.75–7.70 (10H, m), 11.1 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 21.8, 25.6, 30.5, 33.4, 37.4, 41.6, 47.4, 49.8, 56.7, 56.8, 82.0, 128.8, 129.6, 131.3, 131.4, 131.6, 176.9 Anal. Calcd for C24H30ClNO2: C, 72.07; H, 7.56; N, 3.50. Found: C, 72.07; H, 7.53; N, 3.55.
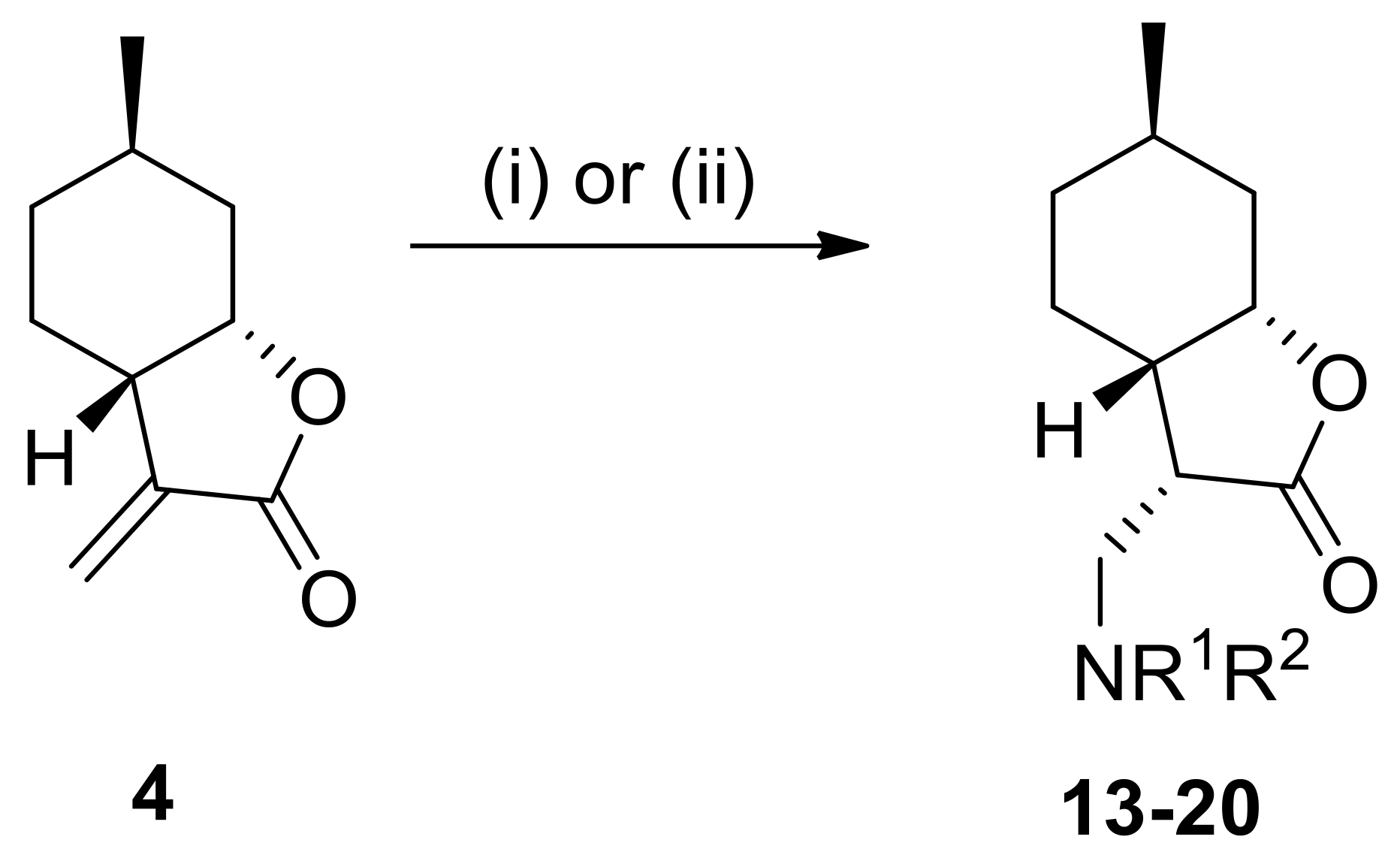
4.3.7. (3S,3aS,6R,7aS)-3-((Benzylamino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one hydrochloride (13)
Prepared from 4 with benzylamine at 25 °C for 20 h. Compound 13 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 60%, white crystals, m.p.: 165–167 °C. = −34.0 (c 0.24, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.73–0.88 (2H, m), 0.88 (1H, d, J = 6.2 Hz), 1.24–1.30 (1H, m), 1.34–1.43 (1H, m), 1.58 (1H, d, J = 12.4 Hz), 1.81 (1H, t, J = 6.2 Hz), 2.06 (1H, d, J = 14.3 Hz), 2.56–2.59 (1H, m), 3.00 (1H, t, J = 10.7 Hz), 3.16 (1H, d, J = 12.0 Hz), 2.42–2.43 (1H, m), 4.20 (1H, q, J = 13.0 Hz), 4.58 (1H, s), 7.42–7.60 (5H, m), 9.54 (2H, s). 13C NMR (125 MHz, DMSO-d6): δ = 21.7, 22.4, 25.8, 31.1, 35.0, 36.2, 41.6, 44.6, 50.2, 78.1, 128.6, 129.0, 130.2, 131.8, 175.6. Anal. Calcd for C17H24ClNO2: C, 65.90; H, 7.81; N, 4.52. Found: C, 65.95; H, 7.80; N, 4.55.
4.3.8. (3S,3aS,6R,7aS)-6-Methyl-3-((((R)-1-phenylethyl)amino)methyl)hexahydrobenzofuran-2(3H)-one hydrochloride (14)
Prepared from 4 with (R)-α-methylbenzylamine at 25 °C for 20 h. Compound 14 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 65%, white crystals, m.p.: 250–252 °C. = −3.0 (c 0.24, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.72–0.78 (1H, m), 0.83–0.90 (1H, m), 0.87 (3H, d, J = 6.4 Hz), 1.24–1.30 (1H, m), 1.33–1.37 (1H, m), 1.57 (1H, d, J = 12.7 Hz), 1.63 (1H, d, J = 6.7 Hz), 1.82–1.85 (1H, m), 2.04 (H, d, J = 14.1 Hz), 2.61–2.66 (1H, m), 2.79–2.84 (1H, m), 2.91–2.97 (1H, m), 3.29–3.31 (1H, m), 4.44 (1H, q, J = 4.8 Hz), 4.55 (1H, d, J = 2.3 Hz), 7.40–7.64 (5H, m), 9.49 (1H, br s), 9.94 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 19.3, 21.7, 22.4, 25.8, 31.1, 35.0, 36.3, 40.4, 44.8, 57.7, 78.1, 127.9, 128.9, 137.0, 175.6. Anal. Calcd for C18H26ClNO2: C, 66.76; H, 8.09; N, 4.32. Found: C, 66.74; H, 8.13; N, 4.35.
4.3.9. (3S,3aS,6R,7aS)-6-Methyl-3-((((S)-1-phenylethyl)amino)methyl)hexahydrobenzofuran-2(3H)-one hydrochloride (15)
Prepared from 4 with (S)-α-methylbenzylamine at 25 °C for 20 h. Compound 15 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 70%, white crystals, m.p.: 230–235 °C. = −57.0 (c 0.21, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.55–0.63 (1H, m), 0.78–0.85 (1H, m), 0.85 (3H, d, J = 6.2 Hz), 1.22–1.30 (2H, m), 1.50 (2H, d, J = 10.9 Hz), 1.64 (1H, d, J = 6.7 Hz), 2.03 (1H, d, J = 13.4 Hz), 2.50–2.56 (1H, m), 2.59–2.63 (1H, m), 3.05 (1H, t, J = 9.7 Hz), 3.42–3.44 (1H, m), 4.47 (1H, br s), 4.57 (1H, d, J = 2.1 Hz), 7.39–7.64 (5H, m), 9.48 (1H, d, J = 7.6 Hz), 10.04 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 19.5, 21.7, 22.1, 25.7, 31.0, 35.0, 36.0, 40.2, 44.5, 57.6, 78.0, 127.8, 128.9, 129.0, 136.9, 175.4. Anal. Calcd for C18H26ClNO2: C, 66.76; H, 8.09; N, 4.32. Found: C, 66.77; H, 8.13; N, 4.29.
4.3.10. (3S,3aS,6R,7aS)-3-((Diethylamino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one hydrochloride (16)
Prepared from 4 with diethylamine at 25 °C for 20 h. Compound 16 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 50%, white crystals, m.p.: 158–163 °C. = −39.0 (c 0.24, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.81–0.93 (1H, m), 0.87 (1H, d, J = 9.0 Hz), 1.20–1.26 (6H, m), 1.26–1.29 (1H, m), 1.33–1.43 (1H, m), 1.60 (1H, d, J = 11.5 Hz), 1.82–1.85 (1H, m), 2.07 (1H, d, J = 14.6 Hz), 2.64–2.66 (1H, m), 3.14–3.22 (6H, m), 3.58 (1H, q, J = 4.2 Hz), 4.58 (1H, d, J = 2.2 Hz), 10.6 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 8.0, 8.6, 21.7, 22.6, 25.8, 31.1, 35.1, 37.1, 42.9, 45.9, 46.4, 47.1, 78.0, 176.1. Anal. Calcd for C14H26ClNO2: C, 60.96; H, 9.50; N, 5.08. Found: C, 60.73; H, 9.53; N, 5.05.
4.3.11. (3S,3aS,6R,7aS)-6-Methyl-3-(piperidin-1-ylmethyl)hexahydrobenzofuran-2(3H)-one hydrochloride (17)
Prepared from 4 with pyridine at 25 °C for 20 h. Compound 17 was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1). Yield: 53%, white crystals, m.p.: 231–233 °C. = −39.0 (c 0.24, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.76–0.81 (1H, m), 0.87–0.92 (1H, m), 0.88 (3H, d, J = 6.0 Hz), 1.22–1.28 (1H, m), 1.31–1.44 (2H, m), 1.59–1.89 (7H, m), 2.07 (1H, d, J = 14.5 Hz), 2.65 (1H, d, J = 5.6 Hz), 2.87–2.97 (2H, m), 3.17–3.22 (2H, m), 3.41 (1H, d, J = 10.9 Hz), 3.52 (1H, d, J = 11.1 Hz), 3.61 (1H, br s), 4.56 (1H, br s), 10.6 (1H, br s) 13C NMR (125 MHz, DMSO-d6): δ = 21.2, 21.7, 22.1, 22.2, 22.8, 25.8, 31.1, 35.1, 37.3, 43.2, 51.2, 51.7, 52.6, 77.9, 176.0. Anal. Calcd for C15H26ClNO2: C, 62.59; H, 9.10; N, 4.87. Found: C, 62.57; H, 9.05; N, 4.93.
4.3.12. (3S,3aS,6R,7aS)-3-((Dibenzylamino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one hydrochloride (18)
Prepared from 4 with dibenzylamine at 70 °C for 72 h. Compound 18 was purified by column chromatography on silica gel (n-hexane/ethyl acetate = 9:1). Yield: 50%, white crystals, m.p.: 118–120 °C. = −23.0 (c 0.21, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.45–0.52 (1H, m), 0.73–0.85 (1H, m), 0.83 (1H, d, J = 6.1 Hz), 1.17–1.30 (3H, m), 1.41 (1H, d, J = 12.4 Hz), 2.03 (1H, d, J = 13.7 Hz), 2.70 (1H, d, J = 4.9 Hz), 2.80 (1H, t, J = 10.2 Hz), 3.20–3.24 (1H, m), 3.78 (1H, br s), 4.35–4.40 (2H, m), 4.49–4.53 (2H, m), 7.33–7.70 (10H, m), 11.3 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 21.6, 22.1, 25.6, 31.0, 35.0, 36.9, 42.8, 46.0, 56.1, 56.6, 77.9, 128.8, 129.1, 129.6, 129.7, 129.9, 131.6, 131.9, 175.6. Anal. Calcd for C24H30ClNO2: C, 72.07; H, 7.56; N, 3.50. Found: C, 72.09; H, 7.53; N, 3.55.
4.4. General Procedure for Nucleophilic Addition of α-Methylene-γ-Butyrolactone with Amino Esters
To the solution of α-methylene-γ-butyrolactone 2 or 4 (1.2 mmol) in dry EtOH (2.0 mL) was added l- or β-alanine ethyl ester hydrochloride (2.4 mmol) and Et3N (2.4 mmol). The reaction mixture was stirred at the appropriate temperature for 20 h. When the reaction was complete (indicated by TLC), EtOH was removed under reduced pressure. The crude residue was purified by column chromatography on silica gel with a mixture of CHCl3 and MeOH (19:1). After solvent evaporation, the addition of a few drops of HCl/EtOH, and recrystallization in diethyl ether, compounds 11 and 12, as well as 19 and 20, respectively, were isolated.
4.4.1. Ethyl 3-(((3R,3aS,6R,7aR)-6-methyl-2-oxooctahydrobenzofuran-3-yl)methylamino) propanoate hydrochloride (11)
Prepared from 2 with β-alanine ethyl ester hydrochloride at 25 °C. Yield: 60%, white crystals, m.p.: 125–135 °C. = −3.7 (c 0.32, MeOH). 1H NMR (500 MHz, CDCl3): δ = 1.00–1.07 (1H, m), 1.02 (3H, d, J = 6.6 Hz), 1.20–1.30 (1H, m), 1.29 (3H, t, J = 7.1 Hz), 1.44–1.50 (1H, m), 1.64–1.72 (2H, m), 1.82 (1H, d, J = 13.6 Hz), 2.01 (1H, d, J = 10.7 Hz), 2.26 (1H, m), 2.89 (1H, dt, J = 4.3, 18.0 Hz), 3.10–3.38 (6H, m), 4.00 (1H, td, J = 3.6, 11.3 Hz), 4.22 (2H, q, J = 7.1 Hz), 8.70 (1H, br s), 11.30 (1H, br s). 13C NMR (125 MHz, CDCl3): δ = 14.2, 22.0, 26.2, 30.2, 31.2, 33.8, 38.1, 42.8, 44.6, 47.2, 48.3, 62.2, 84.0, 171.8, 178.3. Anal. Calcd for C15H26ClNO4: C, 56.33; H, 8.19; N, 4.38. Found: C, 56.35; H, 8.15; N, 4.40.
4.4.2. (S)-Ethyl 2-(((3R,3aS,6R,7aR)-6-methyl-2-oxooctahydrobenzofuran-3-yl)methylamino)propanoate hydrochloride (12)
Prepared from 2 with L-alanine ethyl ester hydrochloride at 25 °C. Yield: 40%, white crystals, m.p.: 150–160 °C. = −3.0 (c 0.28, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.97–1.02 (1H, m), 1.02 (3H, d, J = 6.5 Hz), 1.22 (1H, q, J = 11.4 Hz), 1.33 (3H, t, J = 7.1 Hz), 1.49 (1H, q, J = 11.9 Hz), 1.62–1.66 (2H, m), 1.82 (6H, d, J = 6H, d, J = 6.7 Hz), 1.98 (1H, d, J = 12.4 Hz), 2.27 (1H, d, J = 11.5 Hz), 3.11 (1H, t, J = 10.0 Hz), 3.48–3.48 (1H, m), 3.54 (1H, t, J = 10.0 Hz), 3.96 (1H, q, J = 6.0 Hz), 4.03 (1H, td, J = 3.1, 11.3 Hz), 4.27–4.35 (2H, m), 8.14 (1H, br s), 12.2 (1H, br s). 13C NMR (125 MHz, CDCl3): δ = 14.1, 15.9, 22.0, 26.2, 31.1, 33.9, 38.1, 42.6, 46.3, 48.5, 60.0, 63.3, 83.9, 169.2, 178.6. Anal. Calcd for C15H26ClNO4: C, 56.33; H, 8.19; N, 4.38. Found: C, 56.30; H, 8.20; N, 4.35.
4.4.3. Ethyl 3-(((3S,3aS,6R,7aS)-6-methyl-2-oxooctahydrobenzofuran-3-yl) methylamino)propanoate hydrochloride (19)
Prepared from 4 with β-alanine ethyl ester hydrochloride at 70 °C. Yield: 60%, white crystals, m.p.: 202–205 °C. = −37.0 (c 0.21, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.93 (3H, d, J = 6.4 Hz), 0.92–0.99 (1H, m), 1.09 (1H, q, J = 10.0 Hz), 1.24–1.30 (1H, m), 1.29 (3H, t, J = 6.9 Hz), 1.49–1.62 (1H, m), 1.70 (1H, d, J = 12.3 Hz), 1.78 (1H, d, J = 9.5 Hz), 2.24 (1H, d, J = 14.7 Hz), 2.66 (1H, br s), 2.92 (1H, d, J = 16.7 Hz), 3.13 (1H, d, J = 16.2 Hz), 3.26–3.30 (4H, m), 3.60–3.70 (1H, m), 4.22 (2H, q, J = 7.0 Hz), 4.65 (1H, s), 8.81 (1H, br s), 10.8 (1H, br s). 13C NMR (125 MHz, CDCl3): δ = 14.2, 21.9, 23.3, 26.1, 30.4, 31.5, 35.6, 37.7, 44.4, 44.5, 44.7, 62.0, 79.7, 171.1, 177.7. Anal. Calcd for C15H26ClNO4: C, 56.33; H, 8.19; N, 4.38. Found: C, 56.37; H, 8.18; N, 4.35.
4.4.4. (S)-Ethyl 2-(((3R,3aS,6R,7aR)-6-methyl-2-oxooctahydrobenzofuran-3-yl)methylamino)propanoate hydrochloride (20)
Prepared from 4 with L-alanine ethyl ester hydrochloride at 70 °C. Yield: 44%, white crystals, m.p.: 216–218 °C. = −35.0 (c 0.24, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.92 (3H, d, J = 6.5 Hz), 0.94–1.02 (2H, m), 1.24 (1H, t, J = 12.3 Hz), 1.33 (3H, t, J = 7.0 Hz), 1.50–1.60 (1H, m), 1.70 (1H, d, J = 11.5 Hz), 1.76 (3H, d, J = 6.9 Hz), 1.93 (1H, d, J = 10.6 Hz), 2.00–2.14 (1H, m), 2.23 (1H, d, J = 14.9 Hz), 2.78–2.85 (1H, m), 3.32 (1H, d, J = 17.2 Hz), 3.77 (1H, d, J = 4.5 Hz), 4.08–4.16 (1H, m), 4.30 (2H, q, J = 7.0 Hz), 4.61 (1H, s), 9.80 (1H, br s), 10.37 (1H, br s). 13C NMR (125 MHz, CDCl3): δ = 14.2, 15.0, 22.0, 23.1, 26.1, 31.6, 35.8, 37.7, 41.9, 45.2, 55.8, 63.1, 79.5, 168.7, 176.8. Anal. Calcd for C15H26ClNO4: C, 56.33; H, 8.19; N, 4.38. Found: C, 56.35; H, 8.17; N, 4.40.
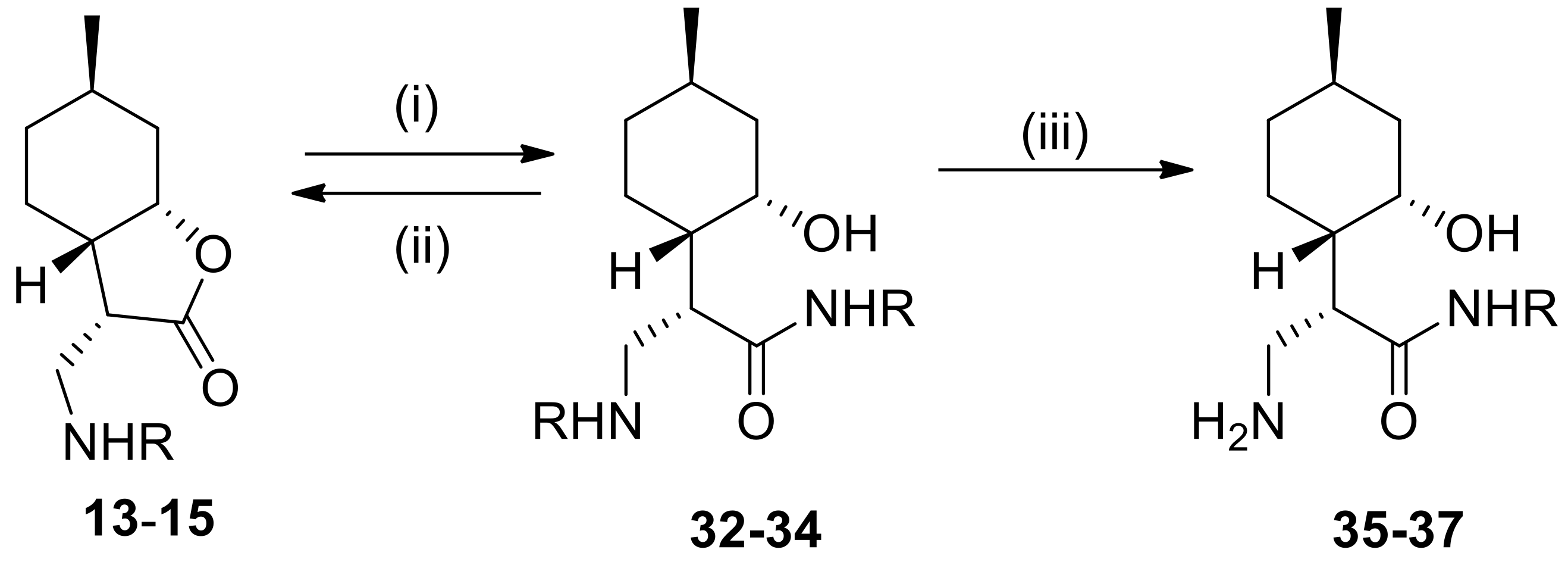
4.5. General Procedure for the Preparation of β-Aminoamides
To a solution of α-methylene-γ-butyrolactone, 2 (1.2 mmol) or β-aminolactones 13–15 (1.2 mmol) in an appropriate solvent (2.0 mL) was added a solution of the appropriate amine (4.8 mmol). The mixture was stirred at the appropriate temperature for 20–72 h. When the reaction was complete (indicated by TLC), the mixture was evaporated to dryness. The crude product was purified by column chromatography on silica gel with CHCl3/MeOH (19:1), resulting in compounds 21–23 and 32–34.
4.5.1. (R)-N-Benzyl-3-(benzylamino)-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)propanamide (21)
Prepared from 2 with benzylamine at 25 °C for 20 h in dry EtOH. Yield: 90%, white crystals, m.p.: 185–195 °C. = −24.0 (c 0.27, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.66–0.73 (1H, m), 0.82–0.92 (2H, m), 0.85 (1H, d, J = 6.5 Hz), 1.34–1.37 (2H, m), 1.52 (1H, d, J = 12.2 Hz), 1.65 (1H, t, J = 11.1 Hz), 1.86 (1H, d, J = 12.4 Hz), 2.93–3.02 (1H, m), 3.24–3.27 (3H, m), 4.06–4.17 (3H, m), 4.44 (1H, dd, J = 6.4, 15.1 Hz), 5.02–5.09 (1H, m), 7.20–7.54 (10H, m), 8.66–8.68 (2H, m), 9.28 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 22.1, 25.1, 30.9, 33.9, 41.7, 42.3, 43.3, 44.4, 47.2, 50.4, 68.5, 126.7, 127.1, 128.2, 128.6, 128.9, 130.0, 131.8, 139.5, 172.0. Anal. Calcd for C24H32N2O2: C, 75.75; H, 8.48; N, 7.36. Found: C, 75.80; H, 8.45; N, 7.35.
4.5.2. (R)-2-((1S,2R,4R)-2-Hydroxy-4-methylcyclohexyl)-N-((R)-1-phenylethyl)-3-(((R)-1-phenylethyl)amino)propanamide (22)
Prepared from 2 with (R)-α-methylbenzylamine at 70 °C for 48 h in dry EtOH. Yield: 58%, colorless oil. = +54.0 (c 0.20, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.72–0.90 (2H, m), 0.89 (1H, 6.4 Hz), 1.04–1.11 (1H, m), 1.25–1.28 (3H, m), 1.36 (3H, d, J = 6.6 Hz), 1.47–1.62 (2H, m), 1.53 (3H, d, J = 6.9 Hz), 1.78–1.87 (2H, m), 1.96 (1H, 1H, J = 13.6 Hz), 2.70 (1H, d, J = 11.8 Hz), 3.00–3.06 (2H, m), 3.42 (1H, d, J = 8.7 Hz), 3.62–3.70 (1H, m), 5.02 (1H, t, J = 7.1 Hz), 7.20–7.49 (10H, m), 8.38 (1H, d, J = 7.1 Hz). 13C NMR (125 MHz, CDCl3): δ = 20.9, 22.0, 22.2, 25.8, 31.7, 34.1, 42.3, 43.5, 44.4, 47.7, 49.8, 58.7, 70.8, 126.7, 127.1, 127.6, 128.5, 128.9, 129.1, 144.4, 172.4. Anal. Calcd for C26H36N2O2: C, 76.43; H, 8.88; N, 6.86. Found: C, 76.45; H, 8.90; N, 6.83.
4.5.3. (R)-2-((1S,2R,4R)-2-Hydroxy-4-methylcyclohexyl)-N-((S)-1-phenylethyl)-3-(((S)-1-phenylethyl)amino)propanamide (23)
Prepared from 2 with (S)-α-methylbenzylamine at 70 °C for 48 h in dry EtOH. Yield: 54%, white crystals, m.p.: 137–148 °C = −55.0 (c 0.26, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.52–0.58 (1H, m), 0.82 (3H, d, J = 6.4 Hz), 0.86–0.91 (2H, m), 1.23–1.37 (2H, m), 1.36 (3H, d, J = 7.1 Hz), 1.56 (3H, d, J = 6.8 Hz), 1.69–1.74 (1H, m), 1.84–1.88 (1H, m), 2.78–2.82 (1H, m), 2.87–2.93 (1H, m), 3.24 (2H, d, J = 10.0 Hz), 4.29 (1H, d, J = 2.3 Hz), 4.91 (1H, quin, J = 7.1 Hz), 5.10 (1H, d, J = 4.9 Hz), 7.15–7.53 (10H, m), 8.30 (1H, d, J = 9.0 Hz), 8.64 (1H, d, J = 7.7 Hz), 9.60 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 19.8, 22.1, 22.3, 24.5, 30.8, 33.7, 41.3, 41.7, 44.2, 47.2, 48.4, 58.0, 68.4, 125.6, 126.4, 127.6, 128.0, 128.7, 128.8, 137.1, 145.4, 171.0. Anal. Calcd for C26H36N2O2: C, 76.43; H, 8.88; N, 6.86. Found: C, 76.40; H, 8.85; N, 6.90.
4.5.4. (S)-N-Benzyl-3-(benzylamino)-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)propanamide (32)
Prepared from 13 with benzylamine at 70 °C for 24 h in dry THF. Yield: 70%, white crystals, m.p.: 251–253 °C. = +21.0 (c = 0.20, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.76–0.80 (1H, m), 0.79 (3H, d, J = 6.3 Hz), 1.00 (1H, t, J = 13.0 Hz), 1.18 (1H, d, J = 10.1 Hz), 1.38–1.43 (1H, m), 1.51–1.58 (2H, m), 1.68 (1H, d, J = 11.2 Hz), 2.75 (1H, t, J = 6.7 Hz), 3.05 (1H, d, J = 11.8 Hz), 3.12–3.22 (1H, m), 3.79 (1H, s), 4.11 (2H, s), 4.18 (1H, dd, J = 5.2, 14.9 Hz), 4.29 (1H, dd, J = 6.1, 14.9 Hz), 4.74 (1H, s), 7.22–7.54 (10H, m), 8.75 (1H, t, J = 5.6 Hz), 9.06 (1H, br s), 9.17 (1H, br s). 13C NMR (125 MHz, DMSO-d6): δ = 22.2, 24.3, 25.1, 34.3, 41.2, 41.8, 42.5, 44.3, 45.3, 50.1, 64.0, 126.8, 127.5, 128.2, 128.6, 128.9, 130.1, 131.7, 139.0, 172.3. Anal. Calcd for C24H32N2O2: C, 75.75; H, 8.48; N, 7.36. Found: C, 75.76; H, 8.50; N, 7.32.
4.5.5. (S)-2-((1S,2S,4R)-2-Hydroxy-4-methylcyclohexyl)-N-((R)-1-phenylethyl)-3-(((R)-1-phenylethyl)amino)propanamide (33)
Prepared from 14 with (R)-α-methylbenzylamine at 70 °C for 72 h in dry THF. Yield: 42%, white crystals, m.p.: 190–192 °C. = +46.0 (c 0.21, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.64–0.72 (1H, m), 0.79 (3H, 6.5 Hz), 0.99 (1H, t, J = 12.5 Hz), 1.14 (1H, d, J = 9.8 Hz), 1.39–1.54 (3H, m), 1.62–1.64 (1H, m), 1.77–1.81 (1H, m), 1.78 (3H, d, J = 6.7 Hz), 2.96 (2H, d, J = 8.6 Hz), 3.27 (1H, d, J = 8.9 Hz), 3.97 (1H, s), 4.36 (1H, q, J = 7.3 Hz), 4.80 (1H, quin, J = 7.3 Hz), 7.17–7.52 (10H, m), 8.23 (1H, d, J = 7.7 Hz) . 13C NMR (125 MHz, CDCl3): δ = 20.2, 22.3, 25.1, 26.0, 34.3, 42.1, 42.6, 45.8, 46.1, 49.7, 59.8, 65.7, 126.6, 127.1, 127.6, 128.5, 129.6, 129.7, 135.7, 143.8, 171.6. Anal. Calcd for C26H36N2O2: C, 76.43; H, 8.88; N, 6.86. Found: C, 76.41; H, 8.85; N, 6.90.
4.5.6. (S)-2-((1S,2S,4R)-2-Hydroxy-4-methylcyclohexyl)-N-((S)-1-phenylethyl)-3-(((S)-1-phenylethyl)amino)propanamide (34)
Prepared by 15 with (S)-α-methylbenzylamine at 70 °C for 72 h in dry THF. Yield: 45%, colorless oil. = −36.0 (c 0.23, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.84 (3H, d, J = 6.3 Hz), 0.83–0.89 (2H, m), 1.01 (1H, t, J = 12.5 Hz), 1.22–1.29 (3H, m), 1.37–1.43 (1H, m), 1.42 (3H, d, J = 6.6 Hz), 1.49 (3H, d, J = 6.9 Hz), 1.51–1.57 (2H, m), 1.66–1.80 (4H, m), 2.61 (1H, br s), 2.77 (2H, d, J = 5.0 Hz), 3.70 (1H, t, J = 6.9 Hz), 3.77 (1H, s), 5.08 (1H, d, J = 7.1 Hz), 7.23–7.41 (10H, m), 7.65 (1H, br s). 13C NMR (125 MHz, CDCl3): δ = 22.0, 22.1, 22.3, 25.2, 25.9, 29.8, 34.7, 42.0, 42.8, 46.5, 47.8, 49.2, 58.9, 66.3, 126.3, 126.5, 127.3, 128.5, 128.7, 128.8, 129.0, 143.7, 172.9. Anal. Calcd for C26H36N2O2: C, 76.43; H, 8.88; N, 6.86. Found: C, 76.45; H, 8.83; N, 6.87.
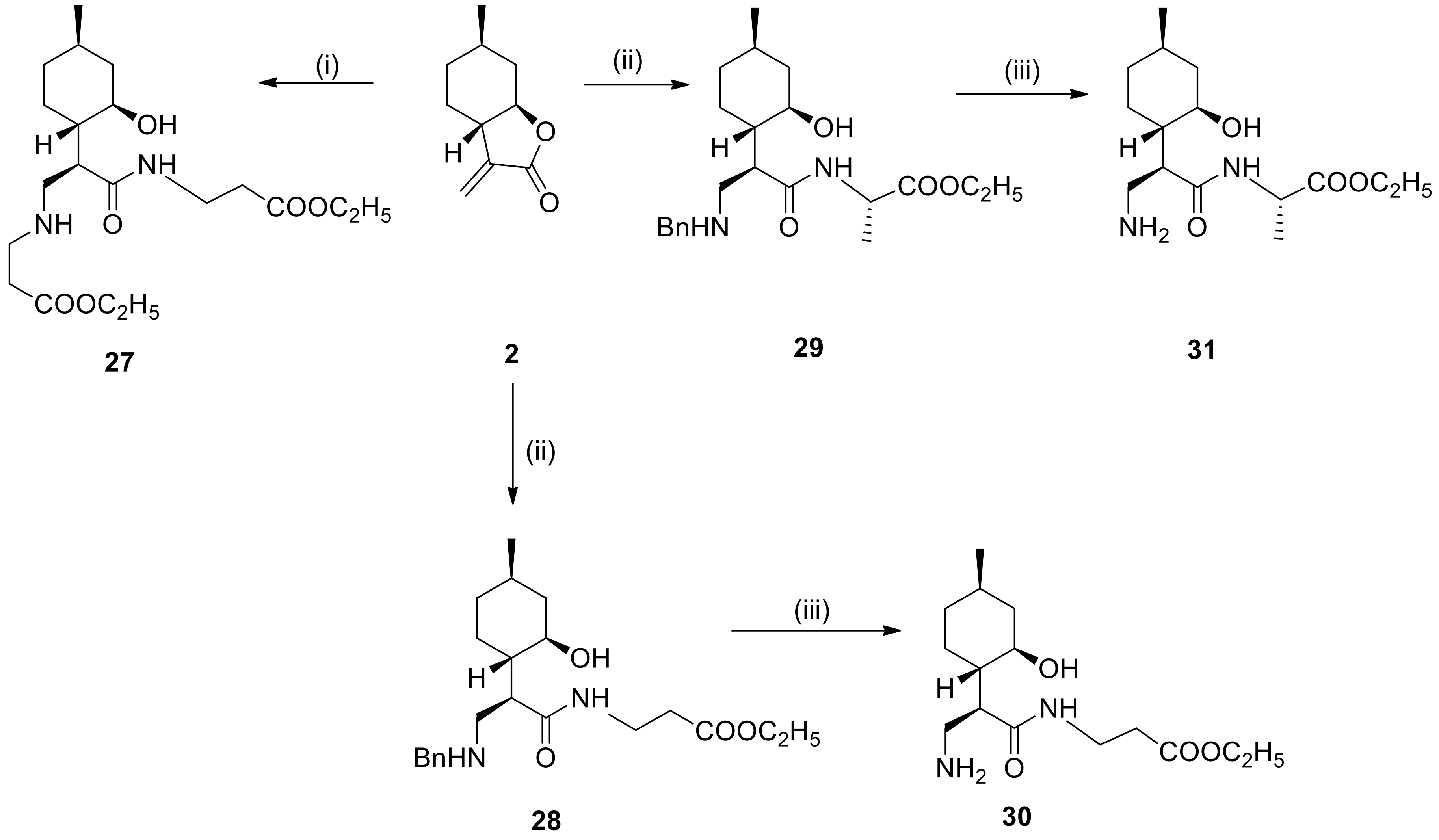
4.7. General Procedure for Preparation of Dipeptides
To the solution of α-methylene-γ-butyrolactone 2 or N-benzyl aminolactone 5a (1.2 mmol) in dry EtOH (2.0 mL) was added L- or β-alanine ethyl ester (3.6 mmol). The mixture was stirred at the appropriate temperature for 48 h. When the reaction was complete (monitored by TLC), the mixture was evaporated to dryness, then purified by column chromatography on silica gel (CHCl3/MeOH = 19:1), affording compounds 27–29.
4.7.1. Ethyl 3-((R)-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)-3-((3-ethoxy-3-oxopropyl)amino)propanemido)propanoate (27)
Prepared from 2 with β-alanine ethyl ester at 25 °C. Yield: 63%, colorless oil. = −14.2 (c 0.33, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.81–1.09 (5H, m), 0.90 (3H, d, J = 6.2 Hz), 1.25–1.28 (11H, m), 1.33–1.40 (1H, m), 1.60 (2H, t, J = 14.6 Hz), 1.72 (1H, t, J = 10.2 Hz), 2.00 (1H, d, J = 13.2 Hz), 2.59 (2H, t, J = 6.3 Hz), 0.83 (1H, d, J = 17.6 Hz), 2.98–3.02 (1H, m), 3.18–3.37 (5H, m), 3.49–3.60 (3H, m), 4.14 (2H, q, J = 7.2 Hz), 4.19 (2H, q, J = 6.4 Hz). 13C NMR (125 MHz, CDCl3): δ = 14.1, 14.2, 22.0, 25.3, 29.7, 30.3, 31.8, 34.1, 34.2, 35.4, 42.1, 43.9, 44.0, 45.1, 47.8, 60.7, 61.6, 70.0, 170.8, 172.3, 173.2. Anal. Calcd for C20H36N2O6: C, 59.98; H, 9.06; N, 6.99. Found: C, 60.00; H, 9.05; N, 6.95.
4.7.2. Ethyl 3-((R)-3-(benzylamino)-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)propanamido)propanoate (28)
Prepared from 5a with β-alanine ethyl ester at 70 °C. Yield: 45%, white crystals, m.p.: 169–173 °C. = −24.0 (c 0.24, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.78–0.98 (3H, m), 0.85 (3H, d, J = 6.4 Hz), 1.24 (3H, t, J = 7.1 Hz), 1.23–1.29 (1H, m), 1.58 (2H, d, J = 10.6 Hz), 1.71 (2H, t, J = 11.5 Hz), 2.57 (2H, t, J = 5.7 Hz), 3.22–3.33 (3H, m), 3.42–3.47 (1H, m), 3.52–3.60 (2H, m), 4.08 (3H, q, J = 7.1 Hz), 4.31–4.34 (1H, m), 7.40 (3H, 6.0 Hz), 7.57 (2H, d, J = 5.3 Hz), 7.86 (1H, br s), 8.18 (1H, t, J = 5.5 Hz).10.08 (1H, br s). 13C NMR (125 MHz, CDCl3): δ = 14.3, 22.0, 25.4, 31.8, 34.0, 34.3, 35.6, 42.3, 43.9, 44.7, 47.8, 52.3, 60.9, 70.2, 129.3, 129.7, 130.1, 130.7, 172.8, 173.4. Anal. Calcd for C22H34N2O4: C, 67.66; H, 8.78; N, 7.17. Found: C, 67.70; H, 8.75; N, 7.20.
4.7.3. (S)-Ethyl 2-((R)-3-(benzylamino)-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)propanamido)propanoate (29)
Prepared from 5a with L-alanine ethyl ester at 70 °C. Yield: 40%, white crystals, m.p.: 115–117 °C. = −30.0 (c 0.25, MeOH). 1H NMR (500 MHz, CDCl3): δ = 0.81–0.89 (2H, m), 0.88 (3H, d, J = 6.5 Hz), 1.04 (1H, q, J = 11.8 Hz), 1.25 (3H, t, J = 7.1 Hz), 1.28–1.38 (1H, m), 1.47 (3H, d, J = 7.2 Hz), 1.58–1.66 (2H, m), 1.81–1.90 (2H, m), 1.95–2.17 (2H, m), 3.10–3.18 (1H, m), 3.27–3.34 (2H, m), 3.64 (1H, d, J = 10.2 Hz), 4.00–4.03 (1H, m), 4.14–4.18 (3H, m), 4.42 (1H, quin, J = 6.9 Hz), 7.39 (3H, d, J = 3.6 Hz), 7.51 (2H, d, J = 3.8 Hz), 7.99 (1H, d, J = 6.1 Hz), 10.1 (br s). 13C NMR (125 MHz, CDCl3): δ = 14.3, 17.3, 22.1, 25.2, 31.8, 34.1, 42.1, 43.8, 44.2, 48.0, 49.0, 52.3, 52.3, 61.3, 70.0, 129.4, 129.8, 130.1, 130.4, 173.0, 173.2. Anal. Calcd for C22H34N2O4: C, 67.66; H, 8.78; N, 7.17. Found: C, 67.65; H, 8.80; N, 7.15.
4.8. General Procedure for Debenzylation
To a suspension of 5% Pd/C or Pd(OH)2/C (100 mg) in MeOH (10 mL) was added β-aminoamides 21–23 and 32–34 or N-benzyldipepetides 28–29 (0.38 mmol) in MeOH (10 mL). The mixture was stirred under H2 at room temperature and normal pressure. When the reaction was complete (indicated by TLC), the mixture was filtered through a Celite pad, the solution was evaporated to dryness and purified by recrystallization in diethyl ether providing 24–26 and 35–37 as well as 30–31, respectively.
4.8.1. (R)-3-Amino-N-benzyl-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)propanamide (24)
Prepared from 21 with 5% Pd/C for 96 h. Yield: 80%, white crystals, m.p.: 226–230 °C. = −24.0 (c 0.29, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.67–0.74 (1H, m), 0.82–0.97 (2H, m), 0.85 (3H, d, J = 6.4 Hz), 1.34–1.37 (2H, m), 1.54 (1H, d, J = 12.0 Hz), 1.69 (1H, t, J = 12.3 Hz), 1.87 (1H, d, J = 11.9 Hz), 2.81 (1H, dd, J = 1.9, 10.1 Hz), 3.08 (1H, t, J = 11.9 Hz), 3.17 (1H, d, J = 10.3 Hz), 3.28 (1H, td, J = 3.9, 10.4 Hz), 4.18 (1H, dd, J = 5.4, 15.1 Hz), 4.43 (1H, dd, J = 6.4, 15.2 Hz), 7.21–7.32 (5H, m), 7.91 (3H, br s), 8.72 (1H, t, J = 5.8 Hz). 13C NMR (125 MHz, DMSO-d6): δ = 22.1, 25.0, 30.9, 34.1, 35.5, 42.2, 42.6, 44.7, 46.8, 68.7, 126.7, 127.1, 128.2, 139.6, 172.2. Anal. Calcd for C17H26N2O2: C, 70.31; H, 9.02; N, 9.65. Found: C, 70.35; H, 9.05; N, 9.60.
4.8.2. (R)-3-Amino-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)-N-((R)-1-phenylethyl)propanamide (25)
Prepared from 22 with 5% Pd/C for 168 h. Yield: 62%, white crystals, m.p.: 145–150 °C. = +28.7 (c 0.31, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.73–0.79 (1H, m), 0.87 (3H, d, J = 6.4 Hz), 0.90–1.00 (2H, m), 1.34 (3H, d, J = 6.9 Hz), 1.37–1.46 (2H, m), 1.59 (1H, d, J = 12.2 Hz), 1.81 (1H, t, J = 12.3 Hz), 1.88 (1H, d, J = 11.9 Hz), 2.81 (1H, dd, J = 2.6, 12.1 Hz), 3.05 (1H, d, J = 12.0 Hz), 3.14–3.17 (1H, m), 3.28 (1H, td, J = 3.7, 10.3 Hz), 5.01 (1H, quin, J = 7.4 Hz), 7.20–7.40 (5H, m), 8.65 (1H, d, J = 8.3 Hz). 13C NMR (125 MHz, DMSO-d6): δ = 22.1, 22.6, 25.1, 31.0, 34.3, 35.5, 42.5, 44.8, 46.5, 48.0, 68.9, 126.4, 126.6, 128.2, 144.3, 171.4. Anal. Calcd for C18H28N2O2: C, 71.02; H, 9.27; N, 9.20. Found: C, 71.05; H, 9.30; N, 9.15.
4.8.3. (R)-3-Amino-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)-N-((S)-1-phenylethyl)propanamide (26)
Prepared from 23 (0.16 g, 0.39 mmol) with Pd(OH)2/C for 200 h. Yield: 65%, white crystals, m.p.: 150–160 °C. = −37.4 (c 0.31, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.59–0.66 (1H, m), 0.74–0.82 (1H, m), 0.84 (3H, d, J = 6.4 Hz), 0.88–0.97 (1H, m), 1.06 (1H, d, J = 10.8 Hz), 1.22–1.26 (1H, m), 1.30–1.37 (1H, m), 1.36 (3H, d, J = 7.0 Hz), 1.42 (1H, d, J = 12.1 Hz), 1.77–1.87 (2H, m), 2.75 (1H, d, J = 11.8 Hz), 3.00 (1H, d, J = 12.0 Hz), 3.16–3.25 (2H, m), 4.93 (1H, quin, J = 7.3 Hz), 7.18–7.29 (5H, m), 8.77 (1H, d, J = 7.7 Hz). 13C NMR (125 MHz, DMSO-d6): δ = 22.1, 22.4, 24.8, 30.9, 42.1, 44.7, 46.7, 48.3, 68.7, 125.7, 126.5, 128.1, 145.5, 171.4. Anal. Calcd for C18H28N2O2: C, 71.02; H, 9.27; N, 9.20. Found: C, 71.00; H, 9.25; N, 9.23.
4.8.4. Ethyl 3-((R)-3-amino-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)propanamido)propanoate (30)
Prepared from 28 with 5% Pd/C for 24 h. Yield: 50%, colorless oil. = −15.5(c 0.31, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.66–0.73 (1H, m), 0.79–1.05 (2H, m), 0.83 (3H, d, J = 6.2 Hz), 1.17 (3H, t, J = 7.0 Hz), 1.22–1.28 (1H, m), 1.29–1.35 (1H, m) 1.41–1.53 (3H, m), 1.73–1.82 (1H, m), 2.36–2.43 (3H, m), 2.59–2.76 (3H, m), 3.16 (1H, s), 3.19–3.24 (3H, m), 4.04 (2H, q, J = 7.1 Hz). 13C NMR (125 MHz, DMSO-d6): δ = 14.1, 22.2, 26.0, 31.1, 34.0, 34.4, 34.7, 44.8, 46.4, 59.9, 69.3. Anal. Calcd for C15H28N2O2: C, 59.97; H, 9.40; N, 9.33. Found: C, 60.00; H, 9.45; N, 9.30.
4.8.5. (S)-Ethyl 2-((R)-3-amino-2-((1S,2R,4R)-2-hydroxy-4-methylcyclohexyl)propanamido)propanoate (31)
Prepared from 29 with 5% Pd/C for 24 h. Yield: 55%, colorless oil. = −20.0 (c 0.30, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.70–0.77 (1H, m), 0.85 (3H, d, J = 6.4 Hz), 0.84–0.93 (2H, m), 1.15 (3H, t, J = 7.1 Hz), 1.28 (2H, d, J = 7.3 H), 1.36 (1H, br s), 1.47–1.56 (2H, m), 1.74 (1H, t, J = 11.6 Hz), 1.86 (1H, d, J = 12.7 Hz), 2.78 (1H, d, J = 11.1 Hz), 3.04 (1H, t, J = 11.8 Hz), 3.13 (1H, d, J = 10.3 Hz), 3.26 (1H, td, J = 3.6, 10.2 Hz), 4.04 (2H, q, J = 6.9 Hz), 4.26 (1H, quin, J = 7.0 Hz), 8.58 (1H, d, J = 1.3 Hz). 13C NMR (125 MHz, DMSO-d6): δ = 14.0, 16.5, 22.2, 24.8, 30.9, 34.2, 35.4, 42.3, 44.7, 46.5, 48.0, 60.4, 68.7, 172.3, 172.6. Anal. Calcd for C15H28N2O2: C, 59.97; H, 9.40; N, 9.33. Found: C, 59.97; H, 9.38; N, 9.35.
4.8.6. (S)-3-Amino-N-benzyl-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)propanamide (35)
Prepared from 32 with 5% Pd/C for 96 h. Yield: 70%, white crystals, m.p.: 270–275 °C. = +36.0 (c 0.26, MeOH). 1H NMR (500 MHz, DMSO-d6): 0.74–0.86 (1H, m), 0.79 (3H, d, J = 6.4 Hz), 1.01 (1H, t, J = 12.8 Hz), 1.18–1.26 (2H, m), 1.34–1.40 (1H, m), 1.47 (1H, t, J = 11.4 Hz), 1.55 (1H, d, J = 12.5 Hz), 1.69 (2H, d, J = 11.0 Hz), 2.23–2.27 (1H, m), 2.67–2.71 (1H, m), 2.77–2.80 (1H, m), 3.84 (1H, s), 4.22–4.31 (1H, m), 7.20–7.32 (5H, m), 8.39 (1H, t, J = 5.6 Hz). 13C NMR (125 MHz, DMSO-d6): δ = 22.5, 25.1, 25.5, 34.6, 40.8, 41.3, 41.9, 42.3, 50.6, 64.3, 126.6, 127.2, 128.2, 139.9, 174.7. Anal. Calcd for C17H26N2O2: C, 70.31; H, 9.02; N, 9.65. Found: C, 70.29; H, 9.03; N, 9.60.
4.8.7. (S)-3-Amino-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)-N-((R)-1-phenylethyl)propanamide (36)
Prepared from 33 with 5% Pd/C for 240 h. Yield: 70%, white crystals, m.p.: 245–250 °C. = +20.0 (c 0.24, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.65–0.72 (1H, m), 0.78 (3H, d, J = 6.3 Hz), 0.95–1.02 (1H, m), 1.36 (3H, d, J = 7.0 Hz), 1.35–1.39 (1H, m), 1.48–1.82 (2H, m), 1.65 (2H, d, J = 12.2 Hz), 2.58–2.64 (1H, m), 2.93 (1H, d, J = 12.1 Hz), 3.02–3.06 (1H, m), 3.74 (1H, s), 4.60 (1H, s), 4.93 (1H, quin, J = 7.3 Hz), 7.20–7.30 (5H, m), 7.87 (3H, br s), 8.68 (1H, d, J = 7.8 Hz). 13C NMR (125 MHz, DMSO-d6): δ = 22.3, 22.5, 24.4, 25.2, 34.3, 37.9, 42.0, 44.9, 48.3, 63.9, 126.0, 126.6, 128.1, 144.7, 171.7. Anal. Calcd for C18H28N2O2: C, 71.02; H, 9.27; N, 9.20. Found: C, 71.00; H, 9.25; N, 9.25.
4.8.8. (S)-3-Amino-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)-N-((S)-1-phenylethyl)propanamide (37)
Prepared from 34 with Pd(OH)2/C for 300 h. Yield: 52%, white crystals, m.p.: 225–228 °C. = −32.0 (c 0.24, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 0.78–0.85 (1H, m), 0.82 (3H, d, J =.6.3 Hz), 0.96–1.19 (1H, m), 1.02 (3H, d, J = 6.3 Hz), 1.10 (3H, d, J = 6.0 Hz), 1.35 (3H, d, J = 7.0 Hz), 1.44–1.55 (2H, m), 1.61–1.73 (3H, m), 2.64 (1H, br s), 2.92 (1H, d, J = 11.4 Hz), 3.03–3.15 (2H, m), 3.89 (1H, s), 4.96 (1H, quin, J =.7.0 Hz), 7.20–7.36 (5H, m), 8.68 (1H, d, J = 7.5 Hz). 13C NMR (125 MHz, DMSO-d6): δ = 22.3, 22.5, 24.4, 25.3, 34.4, 41.3, 42.1, 48.0, 49.0, 64.1, 126.2, 126.7, 128.2, 144.2, 171.9. Anal. Calcd for C18H28N2O2: C, 71.02; H, 9.27; N, 9.20. Found: C, 70.97; H, 9.30; N, 9.17.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}