Cellular and Molecular Events in the Airway Epithelium Defining the Interaction Between House Dust Mite Group 1 Allergens and Innate Defences


Abstract
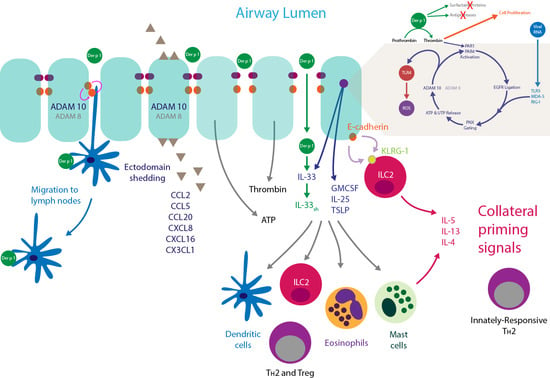
:
1. Introduction
2. Group 1 HDM Allergens: Molecular Characteristics
3. Group 1 HDM Inhalant Allergens and Intercellular Adhesion in the Airway Epithelium
3.1. Tight Junctions and Group 1 HDM Allergens
3.2. Adherens Junctions and Group 1 HDM Allergens
3.3. Desmosomes and Group 1 HDM Allergens
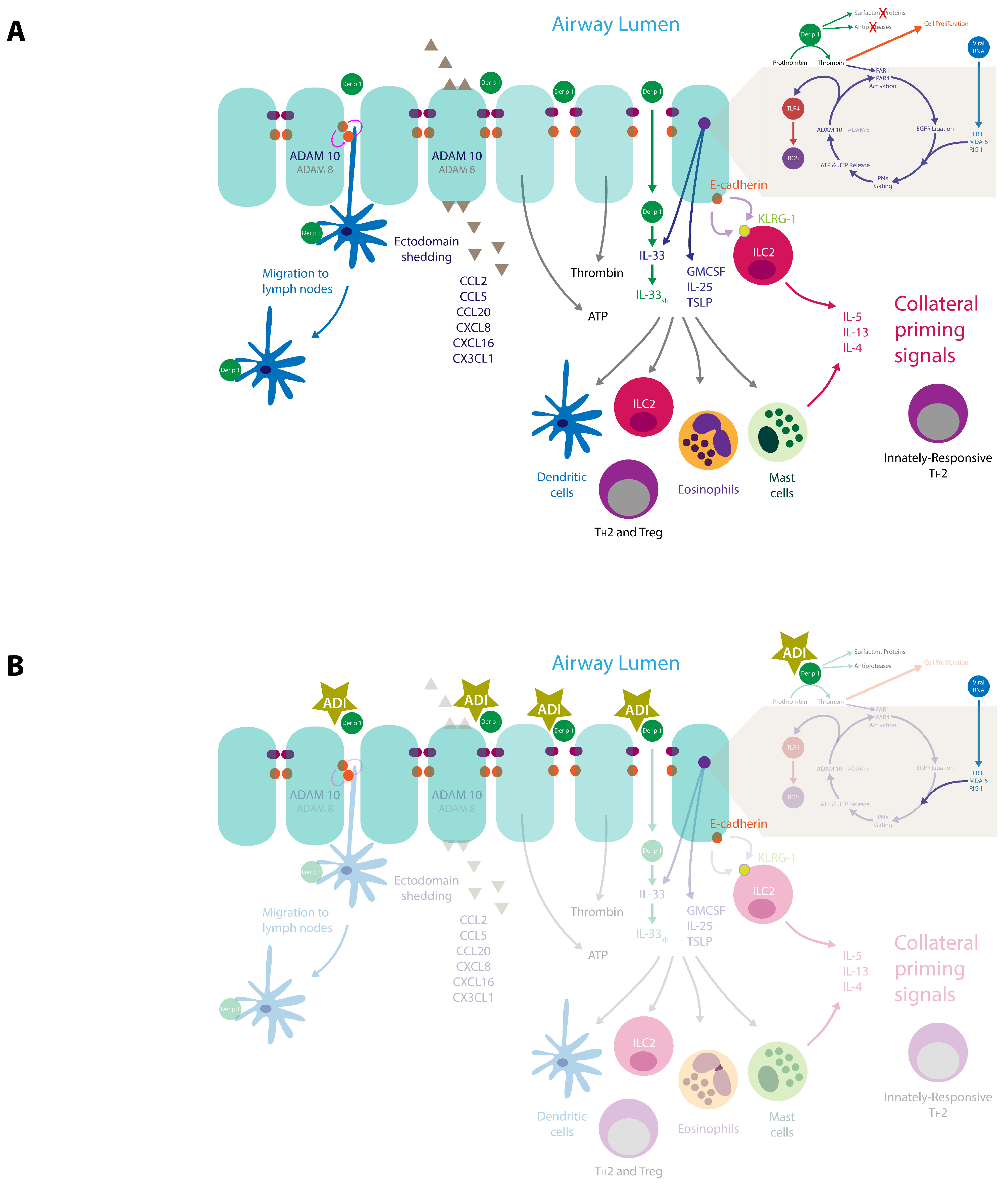
3.4. Consequences of Intercellular Junction Cleavage by HDM Allergens
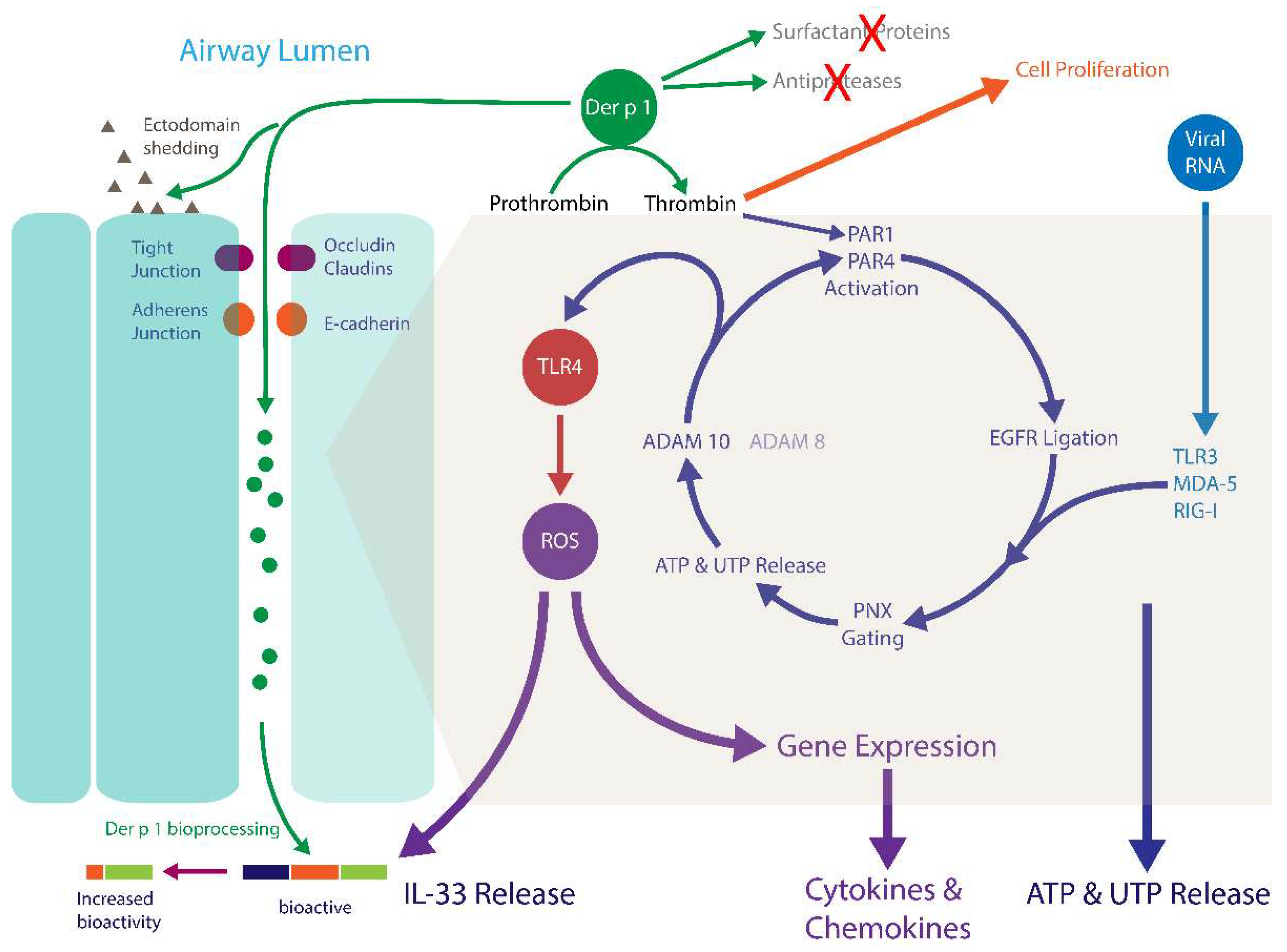
4. Innate Effects of Group 1 HDM Allergens
4.1. Der p 1, PARs, ROS, and Reactive Nitrogen Species (RNS)
4.2. Der p 1, PARs and Epidermal Growth Factor Receptor Signalling
4.3. Der p 1 and Pannexon Gating
4.4. Der p 1 and ADAM 10
4.5. Der p 1 and Toll-Like Receptor 4
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloprotease |
| ADI | allergen delivery inhibitor |
| CCL | C-C chemokine ligand |
| CXCL | C-X-C chemokine ligand |
| DC | dendritic cell |
| Der f | Dermatophagoides farinae |
| Der p | Dermatophagoides pteronyssinus |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| GM-CSF | granulocyte macrophage-colony stimulating factor |
| GPCR | G-protein coupled receptor |
| GUK | guanylate kinase |
| HDM | house dust mite |
| IL | interleukin |
| ILC2 | type 2 innate lymphoid cell |
| IRF-3 | interferon regulatory factor-3 |
| JAK | Janus kinase |
| JAM | junction-associated molecule |
| KLRG-1 | killer cell lectin-like receptor group 1 |
| LPS | lipopolysaccharide |
| MAPK | mitogen activated protein kinase |
| MD-2 | myeloid differentiation protein-2 |
| MDA-5 | melanoma differentiation associated protein-5 |
| MEKK1 | mitogen activated protein kinase kinase kinase I |
| MYLK | myosin light chain kinase |
| NFκB | nuclear factor κB |
| Nrf2 | nuclear factor (erythroid-derived 2)-like 2 |
| PAR | protease-activated receptor |
| PI 3-K | phosphoinositide 3-kinase |
| PNX | pannexon |
| RIG-I | retinoic acid inducible gene-I |
| RLR | retinoic acid inducible gene-I (RIG)-like receptor |
| RNS | reactive nitrogen species |
| ROS | reactive oxidant species |
| RTK | receptor tyrosine kinase |
| SCF | stem cell factor |
| STAT | signal transducer and activation of transcription |
| TAMP | TJ-associated MARVEL domain protein |
| TER | transepithelial electrical resistance |
| Th2 | type 2 T-helper cell |
| TIRAP | toll-interleukin 1 receptor domain-containing adapter protein |
| TJ | tight junction |
| TLR | toll-like receptor |
| TNFα | tumour necrosis factor α |
| TRAM | TRIF-related adapter molecule |
| TRIF | toll-interleukin 1 receptor domain-containing adapter-inducing interferon-β |
| Treg | regulatory T cell |
| TSLP | thymic stromal lymphopoietin |
References
- Matricardi, P.M.; Kleine-Tebbe, J.; Hoffmann, H.J.; Valenta, R.; Hilger, C.; Hofmaier, S.; Aalberse, R.C.; Agache, I.; Asero, R.; Ballmer-Weber, B.; et al. EAACI Molecular Allergology User’s Guide. Pediatr. Allergy Immunol. 2016, 27 (Suppl. 23), 1–250. [Google Scholar] [CrossRef] [PubMed]
- Wickman, M.; Lupinek, C.; Andersson, N.; Belgrave, D.; Asarnoj, A.; Benet, M.; Pinart, M.; Wieser, S.; Garcia-Aymerich, J.; Baar, A.; et al. Detection of IgE Reactivity to a Handful of Allergen Molecules in Early Childhood Predicts Respiratory Allergy in Adolescence. EBioMedicine 2017, 26, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.A.; Richardson, J.P.; Zhang, J.; Robinson, C. The structure and function of allergens. In Middleton’s Allergy—Principles and Practice, 8th Ed.; Adkinson, N.F., Bochner, B.S., Burks, A.W., Busse, W.W., Holgate, S.T., Lemanske, R.F., O’Hehir, R.E., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2014; pp. 398–429. [Google Scholar]
- Dittrich, A.M.; Chen, H.C.; Xu, L.; Ranney, P.; Connolly, S.; Yarovinsky, T.O.; Bottomly, H.K. A new mechanism for inhalational priming: IL-4 bypasses innate immune signals. J. Immunol. 2008, 181, 7307–7315. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, I.; Matha, L.; Steer, C.A.; Ghaedi, M.; Poon, G.F.; Takei, F. Allergen-Experienced Group 2 Innate Lymphoid Cells Acquire Memory-like Properties and Enhance Allergic Lung Inflammation. Immunity 2016, 45, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Huang, Y.; Chen, X.; Hu-Li, J.; Urban, J.F., Jr.; Paul, W.E. Innate immunological function of TH2 cells in vivo. Nat. Immunol. 2015, 16, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, J.; Newton, G.K.; Perrior, T.R.; Robinson, C. Allergen Delivery Inhibitors: A rationale for targeting sentinel innate immune signaling of group 1 house dust mite allergens through structure-based protease inhibitor design. Mol. Pharmacol. 2018, 94, 1007–1030. [Google Scholar] [CrossRef] [PubMed]
- Platts-Mills, T.A.E. Indoor Allergens. In Middleton’s Allergy. Principles and Practice, 7th Ed. ed; Mosby Elsevier: Philadelphia, PA, USA, 2009; pp. 539–555. [Google Scholar]
- Zhang, J.; Chen, J.; Zuo, J.; Newton, G.K.; Perrior, T.R.; Garrod, D.R.; Robinson, C. Allergen Delivery Inhibitors: Characterisation of potent and selective inhibitors of Der p 1 and their attenuation of airway responses to house dust mite allergens in vitro and in vivo. Int. J. Mol. Sci. 2018, 19, 3166. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.K.; Perrior, T.R.; Jenkins, K.; Major, M.R.; Key, R.E.; Stewart, M.R.; Firth-Clark, S.; Lloyd, S.M.; Zhang, J.; Francis-Newton, N.J.; et al. The discovery of potent, selective, and reversible inhibitors of the house dust mite peptidase allergen Der p 1: An innovative approach to the treatment of allergic asthma. J. Med. Chem. 2014, 57, 9447–9462. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.; Zhang, J.; Newton, G.K.; Perrior, T.R. Nonhuman targets in allergic lung conditions. Future Med. Chem. 2013, 5, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hamilton, J.M.; Garrod, D.R.; Robinson, C. Interactions between mature Der p 1 and its free prodomain indicate membership of a new family of C1 peptidases. Allergy 2007, 62, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Meno, K.; Thorsted, P.B.; Ipsen, H.; Kristensen, O.; Larsen, J.N.; Spangfort, M.D.; Gajhede, M.; Lund, K. The crystal structure of recombinant proDer p 1, a major house dust mite proteolytic allergen. J. Immunol. 2005, 175, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Kato, T.; Yasueda, H.; Okumura, K.; Ogawa, H. Analysis of the structure and allergenicity of recombinant pro- and mature Der p 1 and Der f 1: Major conformational IgE epitopes blocked by prodomains. J. Allergy Clin. Immunol. 2005, 115, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Chruszcz, M.; Pomes, A.; Glesner, J.; Vailes, L.D.; Osinski, T.; Porebski, P.J.; Majorek, K.A.; Heymann, P.W.; Platts-Mills, T.A.; Minor, W.; et al. Molecular determinants for antibody binding on group 1 house dust mite allergens. J. Biol. Chem. 2012, 287, 7388–7398. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.C.; Horton, D.; Harrop, R.; Peachell, P.T.; Helm, B.A. Potential allergens stimulate the release of mediators of the allergic response from cells of mast cell lineage in the absence of sensitization with antigen-specific IgE. Eur. J. Immunol. 1996, 26, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Farmer, K.; MacDonald, L.; Kalsheker, N.; Pritchard, D.; Haslett, C.; Lamb, J.; Sallenave, J.M. House dust mite Der p 1 downregulates defenses of the lung by inactivating elastase inhibitors. Am. J. Respir. Cell Mol. Biol. 2003, 29, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, C.R.; Horton, H.; Jones, R.M.; Pritchard, D.I. Heterogeneous proteolytic specificity and activity of the house dust mite proteinase allergen Der p I. Clin. Exp. Allergy 1997, 27, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Robinson, C. Novel method for the purification of house dust mite allergen Der p 1 and its use in structure-based chemical design of novel inhibitors. In Allergy: Methods and Protocols, 2nd Ed.; Lympany, P., Jones, M.G., Eds.; Springer Science & Business Media: New York, NY, USA, 2019. [Google Scholar]
- Ring, P.C.; Wan, H.; Schou, C.; Kroll Kristensen, A.; Roepstorff, P.; Robinson, C. The 18-kDa form of cat allergen Felis domesticus 1 (Fel d 1) is associated with gelatin- and fibronectin-degrading activity. Clin. Exp. Allergy 2000, 30, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The other cells in asthma: Dendritic cell and epithelial cell crosstalk. Curr. Opin. Pulm. Med. 2003, 9, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef] [PubMed]
- van Rijt, L.S.; Lambrecht, B.N. Dendritic cells in asthma: A function beyond sensitization. Clin. Exp. Allergy 2005, 35, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, F.L.; Strickland, D.H.; Thomas, J.A.; Tobagus, I.T.; Napoli, S.; Zosky, G.R.; Turner, D.J.; Sly, P.D.; Stumbles, P.A.; Holt, P.G. Accelerated antigen sampling and transport by airway mucosal dendritic cells following inhalation of a bacterial stimulus. J. Immunol. 2006, 177, 5861–5867. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Kojima, T.; Go, M.; Murata, M.; Ichimiya, S.; Himi, T.; Sawada, N. HLA-DR- and CD11c-positive dendritic cells penetrate beyond well-developed epithelial tight junctions in human nasal mucosa of allergic rhinitis. J. Histochem. Cytochem. 2005, 53, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Nagao, K.; Yokouchi, M.; Sasaki, H.; Amagai, M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J. Exp. Med. 2009, 206, 2937–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalb, T.H.; Chuang, M.T.; Marom, Z.; Mayer, L. Evidence for accessory cell function by class II MHC antigen-expressing airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 1991, 4, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Mezzetti, M.; Soloperto, M.; Fasoli, A.; Mattoli, S. Human bronchial epithelial cells modulate CD3 and mitogen-induced DNA synthesis in T cells but function poorly as antigen-presenting cells compared to pulmonary macrophages. J. Allergy Clin. Immunol. 1991, 87, 930–938. [Google Scholar] [CrossRef]
- Ritz, S.A.; Gajewska, B.U.; Stampfli, M.R.; Jordana, M. Determinants of the immune-inflammatory response in allergic airway inflammation: Overview of antigen presentation and cellular activation. J. Allergy Clin. Immunol. 2000, 106 (Suppl. 5), S206–S212. [Google Scholar] [CrossRef] [PubMed]
- Salik, E.; Tyorkin, M.; Mohan, S.; George, I.; Becker, K.; Oei, E.; Kalb, T.; Sperber, K. Antigen trafficking and accessory cell function in respiratory epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999, 21, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Fromm, M. Claudins and other tight junction proteins. Comp. Physiol. 2012, 2, 1819–1852. [Google Scholar]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Martinez-Estrada, O.M.; Orsenigo, F.; Cordenonsi, M.; Citi, S.; Dejana, E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J. Biol. Chem. 2000, 275, 20520–20526. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Flores-Maldonado, C.; Cereijido, M.; Matter, K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J. Cell. Biochem. 2000, 78, 85–96. [Google Scholar] [CrossRef]
- Balda, M.S.; Whitney, J.A.; Flores, C.; Gonzalez, S.; Cereijido, M.; Matter, K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell. Biol. 1996, 134, 1031–1049. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Winton, H.L.; Soeller, C.; Gruenert, D.C.; Thompson, P.J.; Cannell, M.B.; Stewart, G.A.; Garrod, D.R.; Robinson, C. Quantitative structural and biochemical analyses of tight junction dynamics following exposure of epithelial cells to house dust mite allergen Der p 1. Clin. Exp. Allergy 2000, 30, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Winton, H.L.; Soeller, C.; Tovey, E.R.; Gruenert, D.C.; Thompson, P.J.; Stewart, G.A.; Taylor, G.W.; Garrod, D.R.; Cannell, M.B.; et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J. Clin. Investig. 1999, 104, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Garrod, D.R.; Robinson, C. Novel Der p 1 inhibitors attenuate house dust mite sensitization in mice. Am. J. Crit. Care Med. 2009, 179, A4249. [Google Scholar]
- Shen, L.; Weber, C.R.; Turner, J.R. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J. Cell. Biol. 2008, 181, 683–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.R.; Buschmann, M.M.; Romero-Calvo, I.; Sailer, A.; Shen, L. The role of molecular remodeling in differential regulation of tight junction permeability. Sem. Cell Dev. Biol. 2014, 36, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, B.L.; Mateescu, M.; Patel, A.S.; Wade, K.; Kimura, S.; Gonzales, L.W.; Guttentag, S.; Ballard, P.L.; Koval, M. Developmental regulation of claudin localization by fetal alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1266–L1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, H.; Chiba, S.; Ebina, M.; Furuse, M.; Nukiwa, T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L193–L205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soini, Y. Claudins in lung diseases. Respir. Res. 2011, 12, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweerus, K.; Lachowicz-Scroggins, M.; Gordon, E.; LaFemina, M.; Huang, X.; Parikh, M.; Kanegai, C.; Fahy, J.V.; Frank, J.A. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J. Allergy Clin. Immunol. 2017, 139, 72–81.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, H.; Winton, H.L.; Soeller, C.; Taylor, G.W.; Gruenert, D.C.; Thompson, P.J.; Cannell, M.B.; Stewart, G.A.; Garrod, D.R.; Robinson, C. The transmembrane protein occludin of epithelial tight junctions is a functional target for serine peptidases from faecal pellets of Dermatophagoides pteronyssinus. Clin. Exp. Allergy 2001, 31, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Flynn, A.N.; Itani, O.A.; Moninger, T.O.; Welsh, M.J. Acute regulation of tight junction ion selectivity in human airway epithelia. Proc. Natl. Acad. Sci. USA 2009, 106, 3591–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappi-Blanco, E.; Lehtonen, S.T.; Sormunen, R.; Merikallio, H.M.; Soini, Y.; Kaarteenaho, R.L. Divergence of tight and adherens junction factors in alveolar epithelium in pulmonary fibrosis. Hum. Pathol. 2013, 44, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Niimi, T.; Nagashima, K.; Ward, J.M.; Minoo, P.; Zimonjic, D.B.; Popescu, N.C.; Kimura, S. claudin-18, a novel downstream target gene for the T/EBP/NKX2.1 homeodomain transcription factor, encodes lung- and stomach-specific isoforms through alternative splicing. Mol. Cell. Biol. 2001, 21, 7380–7390. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, C.T.; Farkas, A.E.; Hilgarth, R.S.; Krug, S.M.; Wolf, M.F.; Benedik, J.K.; Fromm, M.; Koval, M.; Parkos, C.; Nusrat, A. Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol. Biol. Cell 2014, 25, 2710–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, S.M.; Schulzke, J.D.; Fromm, M. Tight junction, selective permeability, and related diseases. Sem. Cell Dev. Biol. 2014, 36, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Yeo, N.K.; Jang, Y.J. Rhinovirus infection-induced alteration of tight junction and adherens junction components in human nasal epithelial cells. Laryngoscope 2010, 120, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Balda, M.S.; Matter, K. Signalling at tight junctions during epithelial differentiation and microbial pathogenesis. J. Cell Sci. 2014, 127, 3401–3413. [Google Scholar] [CrossRef] [PubMed]
- Comhair, S.A.; Bhathena, P.R.; Farver, C.; Thunnissen, F.B.; Erzurum, S.C. Extracellular glutathione peroxidase induction in asthmatic lungs: Evidence for redox regulation of expression in human airway epithelial cells. FASEB J. 2001, 15, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, M.; Barajas, B.; Sioutas, C.; Williams, M.A.; Nel, A.E. Nrf2 deficiency in dendritic cells enhances the adjuvant effect of ambient ultrafine particles on allergic sensitization. J. Innate Immunity 2013, 5, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, M.; Anderson, E.L.; Squillace, D.L.; Patil, N.; Maniak, P.J.; Iijima, K.; Kita, H.; O’Grady, S.M. Oxidative stress serves as a key checkpoint for IL-33 release by airway epithelium. Allergy 2017, 72, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Utsch, L.; Folisi, C.; Akkerdaas, J.H.; Logiantara, A.; van de Pol, M.A.; van der Zee, J.S.; Krop, E.J.; Lutter, R.; van Ree, R.; van Rijt, L.S. Allergic sensitization is associated with inadequate antioxidant responses in mice and men. Allergy 2015, 70, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Rangasamy, T.; Bauer, S.M.; Killedar, S.; Karp, M.; Kensler, T.W.; Yamamoto, M.; Breysse, P.; Biswal, S.; Georas, S.N. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J. Immunol. 2008, 181, 4545–4559. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Gajghate, S.; Chatterjee, S.; Mandke, P.; McCormick, S.; Sudini, K.; Kumar, S.; Breysse, P.N.; Diette, G.B.; Sidhaye, V.K.; et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L27–36. [Google Scholar] [CrossRef] [PubMed]
- Tulic, M.K.; Vivinus-Nebot, M.; Rekima, A.; Rabelo Medeiros, S.; Bonnart, C.; Shi, H.; Walker, A.; Dainese, R.; Boyer, J.; Vergnolle, N.; et al. Presence of commensal house dust mite allergen in human gastrointestinal tract: A potential contributor to intestinal barrier dysfunction. Gut 2016, 65, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Vanhook, M.K.; Gambling, T.M.; Carson, J.L.; Boucher, R.C.; Johnson, L.G. Regulation of airway tight junctions by proinflammatory cytokines. Mol. Biol. Cell 2002, 13, 3218–3234. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, E.; Cuzzocrea, S. Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Respir. Res. 2007, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Arlian, L.G.; Morgan, M.S. Immunomodulation of skin cytokine secretion by house dust mite extracts. Int. Arch. Allergy Immunol. 2011, 156, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.K.; Kim, H.J.; Youm, J.K.; Ahn, S.K.; Choi, E.H.; Sohn, M.H.; Kim, K.E.; Hong, J.H.; Shin, D.M.; Lee, S.H. Mite and cockroach allergens activate protease-activated receptor 2 and delay epidermal permeability barrier recovery. J. Investig. Dermatol. 2008, 128, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Hirasawa, Y.; Takai, T.; Mitsuishi, K.; Okuda, M.; Kato, T.; Okumura, K.; Ikeda, S.; Ogawa, H. Reduction of skin barrier function by proteolytic activity of a recombinant house dust mite major allergen Der f 1. J. Investig. Dermatol. 2006, 126, 2719–2723. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Takai, T.; Kato, T.; Kikuchi, Y.; Niyonsaba, F.; Ikeda, S.; Okumura, K.; Ogawa, H. Upregulation of the release of granulocyte-macrophage colony-stimulating factor from keratinocytes stimulated with cysteine protease activity of recombinant major mite allergens, Der f 1 and Der p 1. Int. Arch. Allergy Immunol. 2008, 146, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Oshio, T.; Sasaki, Y.; Funakoshi-Tago, M.; Aizu-Yokota, E.; Sonoda, Y.; Matsuoka, H.; Kasahara, T. Dermatophagoides farinae extract induces severe atopic dermatitis in NC/Nga mice, which is effectively suppressed by the administration of tacrolimus ointment. Int. Immunopharm. 2009, 9, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Brandner, J.M.; Zorn-Kruppa, M.; Yoshida, T.; Moll, I.; Beck, L.A.; De Benedetto, A. Epidermal tight junctions in health and disease. Tissue Barriers 2015, 3, e974451. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Slifka, M.K.; Rafaels, N.M.; Kuo, I.H.; Georas, S.N.; Boguniewicz, M.; Hata, T.; Schneider, L.C.; Hanifin, J.M.; Gallo, R.L.; et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J. Allergy Clin. Immunol. 2011, 128, 242–246.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.F.; Yin, Y.; Runswick, S.K.; Stewart, G.A.; Thompson, P.J.; Garrod, D.R.; Robinson, C. Peptidase allergen Der p 1 initiates apoptosis of epithelial cells independently of tight junction proteolysis. Mol. Membr. Biol. 2003, 20, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. 2000, 19, 2024–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Mariscal, L.; Dominguez-Calderon, A.; Raya-Sandino, A.; Ortega-Olvera, J.M.; Vargas-Sierra, O.; Martinez-Revollar, G. Tight junctions and the regulation of gene expression. Sem. Cell Dev. Biol. 2014, 36, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mrsny, R.J. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J. Cell Biol. 2000, 148, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Steed, E.; Elbediwy, A.; Vacca, B.; Dupasquier, S.; Hemkemeyer, S.A.; Suddason, T.; Costa, A.C.; Beaudry, J.B.; Zihni, C.; Gallagher, E.; et al. MarvelD3 couples tight junctions to the MEKK1-JNK pathway to regulate cell behavior and survival. J. Cell. Biol. 2014, 204, 821–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Bubeck Wardenburg, J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, J.; Mangat, S.C.; Perera Baruhupolage, C.; Garrod, D.R.; Robinson, C. Pathways of airway oxidant formation by house dust mite allergens and viral RNA converge through myosin motors, pannexons and Toll-like receptor 4. Immunity Inflamm. Dis. 2018, 6, 276–296. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, J.; Tachie-Menson, T.; Shukla, N.; Garrod, D.R.; Robinson, C. Allergen-dependent oxidant formation requires purinoceptor activation of ADAM 10 and prothrombin. J. Allergy Clin. Immunol. 2017, 139, 2023–2026.e9. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Yano, M.; Ohki, Y.; Tomura, M.; Nakano, N. Occludin expression in epidermal γδ T cells in response to epidermal stress causes them to migrate into draining lymph nodes. J. Immunol. 2017, 199, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Zimmerli, S.C.; Hauser, C. Langerhans cells and lymph node dendritic cells express the tight junction component claudin-1. J. Investig. Dermatol. 2007, 127, 2381–2390. [Google Scholar] [CrossRef] [PubMed]
- Nawijn, M.C.; Hackett, T.L.; Postma, D.S.; van Oosterhout, A.J.; Heijink, I.H. E-cadherin: Gatekeeper of airway mucosa and allergic sensitization. Trends Immunol. 2011, 32, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Kies, P.M.; Kauffman, H.F.; Postma, D.S.; van Oosterhout, A.J.; Vellenga, E. Down-regulation of E-cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor-dependent Th2 cell-promoting activity. J. Immunol. 2007, 178, 7678–7685. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalsheker, N.A.; Deam, S.; Chambers, L.; Sreedharan, S.; Brocklehurst, K.; Lomas, D.A. The house dust mite allergen Der p1 catalytically inactivates alpha 1-antitrypsin by specific reactive centre loop cleavage: A mechanism that promotes airway inflammation and asthma. Biochim. Biophys. Res. Commun. 1996, 221, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Laing, P.; Sewell, H.F.; Shakib, F. Der p I, a major allergen of the house dust mite, proteolytically cleaves the low-affinity receptor for human IgE (CD23). Eur. J. Immunol. 1995, 25, 3191–3194. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Sewell, H.F.; Shakib, F. Proteolytic cleavage of CD25, the alpha subunit of the human T cell interleukin 2 receptor, by Der p 1, a major mite allergen with cysteine protease activity. J. Exp. Med. 1998, 187, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Duval, A.; Schmitt, P.; Roga, S.; Camus, M.; Stella, A.; Burlet-Schiltz, O.; Gonzalez-de-Peredo, A.; Girard, J.P. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat. Immunol. 2018, 19, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Lefrancais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, E.; Hansen, K.K.; Astudillo Fernandez, O.; Coulon, L.; Bex, F.; Duhant, X.; Jaumotte, E.; Hollenberg, M.D.; Jacquet, A. The house dust mite allergen Der p 1, unlike Der p 3, stimulates the expression of interleukin-8 in human airway epithelial cells via a proteinase-activated receptor-2-independent mechanism. J. Biol. Chem. 2006, 281, 6910–6923. [Google Scholar] [CrossRef] [PubMed]
- Asokananthan, N.; Graham, P.T.; Stewart, D.J.; Bakker, A.J.; Eidne, K.A.; Thompson, P.J.; Stewart, G.A. House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: The cysteine protease allergen, Der p 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. J. Immunol. 2002, 169, 4572–4578. [Google Scholar] [CrossRef] [PubMed]
- Asokananthan, N.; Graham, P.T.; Fink, J.; Knight, D.A.; Bakker, A.J.; McWilliam, A.S.; Thompson, P.J.; Stewart, G.A. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8, and prostaglandin E2 release from human respiratory epithelial cells. J. Immunol. 2002, 168, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Bara, I.; Gilbert, G.; Carvalho, G.; Trian, T.; Ozier, A.; Gillibert-Duplantier, J.; Ousova, O.; Maurat, E.; Thumerel, M.; et al. Protease activated receptor-2 expression and function in asthmatic bronchial smooth muscle. PLoS ONE 2014, 9, e86945. [Google Scholar] [CrossRef] [PubMed]
- De Campo, B.A.; Henry, P.J. Stimulation of protease-activated receptor-2 inhibits airway eosinophilia, hyperresponsiveness and bronchoconstriction in a murine model of allergic inflammation. Br. J. Pharm. 2005, 144, 1100–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebeling, C.; Forsythe, P.; Ng, J.; Gordon, J.R.; Hollenberg, M.; Vliagoftis, H. Proteinase-activated receptor 2 activation in the airways enhances antigen-mediated airway inflammation and airway hyperresponsiveness through different pathways. J. Allergy Clin. Immunol. 2005, 115, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Post, S.; Heijink, I.H.; Petersen, A.H.; de Bruin, H.G.; van Oosterhout, A.J.; Nawijn, M.C. Protease-activated receptor-2 activation contributes to house dust mite-induced IgE responses in mice. PLoS ONE 2014, 9, e91206. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Divanovic, S.; Visintin, A.; Blanchard, C.; Hegde, R.S.; Madan, R.; Thorne, P.S.; Wills-Karp, M.; Gioannini, T.L.; Weiss, J.P.; et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature 2009, 457, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Gough, L.; Schulz, O.; Sewell, H.F.; Shakib, F. The cysteine protease activity of the major dust mite allergen Der p 1 selectively enhances the immunoglobulin E antibody response. J. Exp. Med. 1999, 190, 1897–1902. [Google Scholar] [CrossRef] [PubMed]
- Gough, L.; Sewell, H.F.; Shakib, F. The proteolytic activity of the major dust mite allergen Der p 1 enhances the IgE antibody response to a bystander antigen. Clin. Exp. Allergy 2001, 31, 1594–1598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J.; Allen-Philbey, K.; Perera Baruhupolage, C.; Tachie-Menson, T.; Mangat, S.C.; Garrod, D.R.; Robinson, C. Innate generation of thrombin and intracellular oxidants in airway epithelium by allergen Der p 1. J. Allergy Clin. Immunol. 2016, 138, 1224–1227. [Google Scholar] [CrossRef] [PubMed]
- Arachiche, A.; Mumaw, M.M.; de la Fuente, M.; Nieman, M.T. Protease-activated receptor 1 (PAR1) and PAR4 heterodimers are required for PAR1-enhanced cleavage of PAR4 by alpha-thrombin. J. Biol. Chem. 2013, 288, 32553–32562. [Google Scholar] [CrossRef] [PubMed]
- Brims, F.J.; Chauhan, A.J.; Higgins, B.; Shute, J.K. Coagulation factors in the airways in moderate and severe asthma and the effect of inhaled steroids. Thorax 2009, 64, 1037–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, J.D.; Majoor, C.J.; van ‘t Veer, C.; Bel, E.H.; van der Poll, T. Asthma and coagulation. Blood 2012, 119, 3236–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabazza, E.C.; Taguchi, O.; Tamaki, S.; Takeya, H.; Kobayashi, H.; Yasui, H.; Kobayashi, T.; Hataji, O.; Urano, H.; Zhou, H.; et al. Thrombin in the airways of asthmatic patients. Lung 1999, 177, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Yoshikawa, T. Up-regulation of thrombin activity induced by vascular endothelial growth factor in asthmatic airways. Chest 2007, 132, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Schouten, M.; van de Pol, M.A.; Levi, M.; van de Pol, T.; van der Zee, J.S. Early activation of coagulation after allergen challenge in patients with allergic asthma. J. Thromb. Haemost. 2009, 7, 1592–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terada, M.; Kelly, E.A.; Jarjour, N.N. Increased thrombin activity after allergen challenge: A potential link to airway remodeling? Am. J. Respir. Crit. Care Med. 2004, 169, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Goeijenbier, M.; van Gorp, E.C.; Van den Brand, J.M.; Stittelaar, K.; Bakhtiari, K.; Roelofs, J.J.; van Amerongen, G.; Kuiken, T.; Martina, B.E.; Meijers, J.C.; et al. Activation of coagulation and tissue fibrin deposition in experimental influenza in ferrets. BMC Microbiol. 2014, 14, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniak, S.; Owens, A.P., 3rd; Baunacke, M.; Williams, J.C.; Lee, R.D.; Weithauser, A.; Sheridan, P.A.; Malz, R.; Luyendyk, J.P.; Esserman, D.A.; Trejo, J.; et al. PAR-1 contributes to the innate immune response during viral infection. J. Clin. Investig. 2013, 123, 1310–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groskreutz, D.J.; Monick, M.M.; Powers, L.S.; Yarovinsky, T.O.; Look, D.C.; Hunninghake, G.W. Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to Increased double-stranded RNA responsiveness in airway epithelial cells. J. Immunol. 2006, 176, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Silkoff, P.E.; Flavin, S.; Gordon, R.; Loza, M.J.; Sterk, P.J.; Lutter, R.; Diamant, Z.; Turner, R.B.; Lipworth, B.J.; Proud, D.; et al. Toll-like receptor 3 blockade in rhinovirus-induced experimental asthma exacerbations: A randomized controlled study. J. Allergy Clin. Immunol. 2018, 141, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.K.; Loh, X.Y.; Peh, H.Y.; Tan, W.N.; Tan, W.S.; Li, N.; Tay, I.J.; Wong, W.S.; Engelward, B.P. House dust mite-induced asthma causes oxidative damage and DNA double-strand breaks in the lungs. J. Allergy Clin. Immunol. 2016, 138, 84–96.e1. [Google Scholar] [CrossRef] [PubMed]
- Comhair, S.A.A.; Erzurum, S.C. Redox control of asthma: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 93–124. [Google Scholar] [CrossRef] [PubMed]
- van Rijt, L.S.; Utsch, L.; Lutter, R.; van Ree, R. Oxidative Stress: Promoter of allergic sensitization to protease allergens? Int. J. Mol. Sci. 2017, 18, 1112. [Google Scholar] [CrossRef] [PubMed]
- Fryer, A.A.; Bianco, A.; Hepple, M.; Jones, P.W.; Strange, R.C.; Spiteri, M.A. Polymorphism at the glutathione S-transferase GSTP1 locus. A new marker for bronchial hyperresponsiveness and asthma. Am. J. Respir. Crit. Care Med. 2000, 161, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Mapp, C.E.; Fryer, A.A.; De Marzo, N.; Pozzato, V.; Padoan, M.; Boschetto, P.; Strange, R.C.; Hemmingsen, A.; Spiteri, M.A. Glutathione S-transferase GSTP1 is a susceptibility gene for occupational asthma induced by isocyanates. J. Allergy Clin. Immunol. 2002, 109, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Sackesen, C.; Ercan, H.; Dizdar, E.; Soyer, O.; Gumus, P.; Tosun, B.N.; Buyuktuncer, Z.; Karabulut, E.; Besler, T.; Kalayci, O. A comprehensive evaluation of the enzymatic and nonenzymatic antioxidant systems in childhood asthma. J. Allergy Clin. Immunol. 2008, 122, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, M.A.; Bianco, A.; Strange, R.C.; Fryer, A.A. Polymorphisms at the glutathione S-transferase, GSTP1 locus: A novel mechanism for susceptibility and development of atopic airway inflammation. Allergy 2000, 55 (Suppl. 61), 15–20. [Google Scholar] [CrossRef] [PubMed]
- Babusikova, E.; Jesenak, M.; Evinova, A.; Banovcin, P.; Dobrota, D. Frequency of polymorphism -262 c/t in catalase gene and oxidative damage in Slovak children with bronchial asthma. Arch. Bronconeumol. 2013, 49, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A.V.; Ivanov, V.P.; Bogomazov, A.D.; Freidin, M.B.; Illig, T.; Solodilova, M.A. Antioxidant defense enzyme genes and asthma susceptibility: Gender-specific effects and heterogeneity in gene-gene interactions between pathogenetic variants of the disease. BioMed Res. Int. 2014, 2014, 708903. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.J.; Shamsuddin, M.; Sporn, P.H.; Denenberg, M.; Anderson, J. Reduced superoxide dismutase in lung cells of patients with asthma. Free Radical Biol. Med. 1997, 22, 1301–1307. [Google Scholar] [CrossRef]
- Yucesoy, B.; Johnson, V.J.; Lummus, Z.L.; Kissling, G.E.; Fluharty, K.; Gautrin, D.; Malo, J.L.; Cartier, A.; Boulet, L.P.; Sastre, J.; et al. Genetic variants in antioxidant genes are associated with diisocyanate-induced asthma. Toxicol. Sci. 2012, 129, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Cao, W.; Kasturi, S.P.; Ravindran, R.; Nakaya, H.I.; Kundu, K.; Murthy, N.; Kepler, T.B.; Malissen, B.; Pulendran, B. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat. Immunol. 2010, 11, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Vieira, R.P.; Grimm, M.; Durk, T.; Cicko, S.; Zeiser, R.; Jakob, T.; Martin, S.F.; Blumenthal, B.; Sorichter, S.; et al. A potential role for P2X7R in allergic airway inflammation in mice and humans. Am. J. Respir. Cell Mol. Biol. 2011, 44, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Hammad, H.; van Nimwegen, M.; Kool, M.; Willart, M.A.; Muskens, F.; Hoogsteden, H.C.; Luttmann, W.; Ferrari, D.; Di Virgilio, F.; et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat. Med. 2007, 13, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Kouzaki, H.; Iijima, K.; Kobayashi, T.; O’Grady, S.M.; Kita, H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J. Immunol. 2011, 186, 4375–4387. [Google Scholar] [CrossRef] [PubMed]
- Basoglu, O.K.; Pelleg, A.; Essilfie-Quaye, S.; Brindicci, C.; Barnes, P.J.; Kharitonov, S.A. Effects of aerosolized adenosine 5′-triphosphate vs adenosine 5′-monophosphate on dyspnea and airway caliber in healthy nonsmokers and patients with asthma. Chest 2005, 128, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Schulman, E.S.; Glaum, M.C.; Post, T.; Wang, Y.; Raible, D.G.; Mohanty, J.; Butterfield, J.H.; Pelleg, A. ATP modulates anti-IgE-induced release of histamine from human lung mast cells. Am. J. Respir. Cell Mol. Biol. 1999, 20, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Boitano, S.; Isakson, B.E.; Evans, W.H. Communication of calcium waves in airway epithelial cells. Mol. Biol. Cell 2000, 11, 328a. [Google Scholar]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Misaghi, S.; Newton, K.; Gilmour, L.L.; Louie, S.; Cupp, J.E.; Dubyak, G.R.; Hackos, D.; Dixit, V.M. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J. Immunol. 2011, 186, 6553–6561. [Google Scholar] [CrossRef] [PubMed]
- Sandilos, J.K.; Chiu, Y.H.; Chekeni, F.B.; Armstrong, A.J.; Walk, S.F.; Ravichandran, K.S.; Bayliss, D.A. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J. Biol. Chem. 2012, 287, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010, 6, e1001178. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.G.; Oh, S.Y.; Park, H.K.; Kim, Y.S.; Shim, E.J.; Lee, H.S.; Oh, M.H.; Bang, B.; Chun, E.Y.; Kim, S.H.; et al. TH2 and TH1 lung inflammation induced by airway allergen sensitization with low and high doses of double-stranded RNA. J. Allergy Clin. Immunol. 2007, 120, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Weskamp, G.; Ford, J.W.; Sturgill, J.; Martin, S.; Docherty, A.J.; Swendeman, S.; Broadway, N.; Hartmann, D.; Saftig, P.; Umland, S.; et al. ADAM10 is a principal ‘sheddase’ of the low-affinity immunoglobulin E receptor CD23. Nat. Immunol. 2006, 7, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Gough, P.J.; Garton, K.J.; Wille, P.T.; Rychlewski, M.; Dempsey, P.J.; Raines, E.W. A disintegrin and metalloproteinase 10-mediated cleavage and shedding regulates the cell surface expression of CXC chemokine ligand 16. J. Immunol. 2004, 172, 3678–3685. [Google Scholar] [CrossRef] [PubMed]
- Post, S.; Rozeveld, D.; Jonker, M.R.; Bischoff, R.; van Oosterhout, A.J.; Heijink, I.H. ADAM10 mediates the house dust mite-induced release of chemokine ligand CCL20 by airway epithelium. Allergy 2015, 70, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Saftig, P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: Physiological and cellular functions. Sem. Cell Dev. Biol. 2009, 20, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Faber, T.W.; Pullen, N.A.; Fernando, J.F.; Kolawole, E.M.; McLeod, J.J.; Taruselli, M.; Williams, K.L.; Rivera, K.O.; Barnstein, B.O.; Conrad, D.H.; et al. ADAM10 is required for SCF-induced mast cell migration. Cell. Immunol. 2014, 290, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooley, L.F.; Martin, R.K.; Zellner, H.B.; Irani, A.M.; Uram-Tuculescu, C.; El Shikh, M.E.; Conrad, D.H. Increased B Cell ADAM10 in allergic patients and Th2 prone mice. PLoS ONE 2015, 10, e0124331. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, E.; Crahay, C.; Garbacki, N.; Hennuy, B.; Gueders, M.; Noel, A.; Foidart, J.M.; Grooten, J.; Colige, A.; Piette, J.; et al. New asthma biomarkers: Lessons from murine models of acute and chronic asthma. Am. J. Physiol. Lung Cell. Mol. Biol. 2009, 296, L185–L197. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.; Soong, G.; Sokol, S.; Reddy, B.; Gomez, M.I.; Van Heeckeren, A.; Prince, A. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Dabbagh, K.; Dahl, M.E.; Stepick-Biek, P.; Lewis, D.B. Toll-like receptor 4 is required for optimal development of Th2 immune responses: Role of dendritic cells. J. Immunol. 2002, 168, 4524–4530. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.C.; Piggott, D.A.; Huleatt, J.W.; Visintin, I.; Herrick, C.A.; Bottomly, K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 2002, 196, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Oh, S.Y.; Jeon, S.G.; Park, H.W.; Lee, S.Y.; Chun, E.Y.; Bang, B.; Lee, H.S.; Oh, M.H.; Kim, Y.S.; et al. Airway exposure levels of lipopolysaccharide determine type 1 versus type 2 experimental asthma. J. Immunol. 2007, 178, 5375–5382. [Google Scholar] [CrossRef] [PubMed]
- Nigo, Y.I.; Yamashita, M.; Hirahara, K.; Shinnakasu, R.; Inami, M.; Kimura, M.; Hasegawa, A.; Kohno, Y.; Nakayama, T. Regulation of allergic airway inflammation through Toll-like receptor 4-mediated modification of mast cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2286–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piggott, D.A.; Eisenbarth, S.C.; Xu, L.; Constant, S.L.; Huleatt, J.W.; Herrick, C.A.; Bottomly, K. MyD88-dependent induction of allergic Th2 responses to intranasal antigen. J. Clin. Investig. 2005, 115, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redecke, V.; Hacker, H.; Datta, S.K.; Fermin, A.; Pitha, P.M.; Broide, D.H.; Raz, E. Cutting edge: Activation of Toll-like receptor 2 induces a Th2 immune response and promotes experimental asthma. J. Immunol. 2004, 172, 2739–2743. [Google Scholar] [CrossRef] [PubMed]
- Ritz, S.A.; Cundall, M.J.; Gajewska, B.U.; Alvarez, D.; Gutierrez-Ramos, J.C.; Coyle, A.J.; McKenzie, A.N.; Stampfli, M.R.; Jordana, M. Granulocyte macrophage colony-stimulating factor-driven respiratory mucosal sensitization induces Th2 differentiation and function independently of interleukin-4. Am. J. Respir. Cell Mol. Biol. 2002, 27, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Chieppa, M.; Perros, F.; Willart, M.A.; Germain, R.N.; Lambrecht, B.N. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 2009, 15, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.A.; Loh, Z.; Gan, W.J.; Zhang, V.; Yang, H.; Li, J.H.; Yamamoto, Y.; Schmidt, A.M.; Armour, C.L.; Hughes, J.M.; et al. Receptor for advanced glycation end products and its ligand high-mobility group box-1 mediate allergic airway sensitization and airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Willart, M.A.; Deswarte, K.; Pouliot, P.; Braun, H.; Beyaert, R.; Lambrecht, B.N.; Hammad, H. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J. Exp. Med. 2012, 209, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- McAlees, J.W.; Whitehead, G.S.; Harley, I.T.; Cappelletti, M.; Rewerts, C.L.; Holdcroft, A.M.; Divanovic, S.; Wills-Karp, M.; Finkelman, F.D.; Karp, C.L.; et al. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol. 2015, 8, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Lee, J.E.; Lim, H.; Shin, H.W.; Khalmuratova, R.; Choi, G.; Kim, H.S.; Choi, W.S.; Park, Y.J.; Shim, I.; et al. Fibrinogen cleavage products and Toll-like receptor 4 promote the generation of programmed cell death 1 ligand 2-positive dendritic cells in allergic asthma. J. Allergy Clin. Immunol. 2018, 142, 530–541.e6. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C. Endogenous ligands of TLR2 and TLR4: Agonists or assistants? J. Leuk. Biol. 2010, 87, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.P.; Patel, K.; Ye, S. Functional Toll-like receptor 4 mutations modulate the response to fibrinogen. Thromb. Haemost. 2008, 100, 301–307. [Google Scholar] [PubMed]
- Millien, V.O.; Lu, W.; Shaw, J.; Yuan, X.; Mak, G.; Roberts, L.; Song, L.Z.; Knight, J.M.; Creighton, C.J.; Luong, A.; et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science 2013, 341, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadiz, G.; Sporn, L.A.; Goss, R.A.; Lawrence, S.O.; Marder, V.J.; Simpson-Haidaris, P.J. Polarized secretion of fibrinogen by lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 1997, 17, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Musso, T.; Gusella, G.L.; Brooks, A.; Longo, D.L.; Varesio, L. Interleukin-4 inhibits indoleamine 2,3-dioxygenase expression in human monocytes. Blood 1994, 83, 1408–1411. [Google Scholar] [PubMed]
- Saluzzo, S.; Gorki, A.D.; Rana, B.M.J.; Martins, R.; Scanlon, S.; Starkl, P.; Lakovits, K.; Hladik, A.; Korosec, A.; Sharif, O.; et al. First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep. 2017, 18, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- De Kleer, I.M.; Kool, M.; de Bruijn, M.J.; Willart, M.; van Moorleghem, J.; Schuijs, M.J.; Plantinga, M.; Beyaert, R.; Hams, E.; Fallon, P.G.; et al. Perinatal activation of the interleukin-33 pathway pPromotes type 2 immunity in the developing lung. Immunity 2016, 45, 1285–1298. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
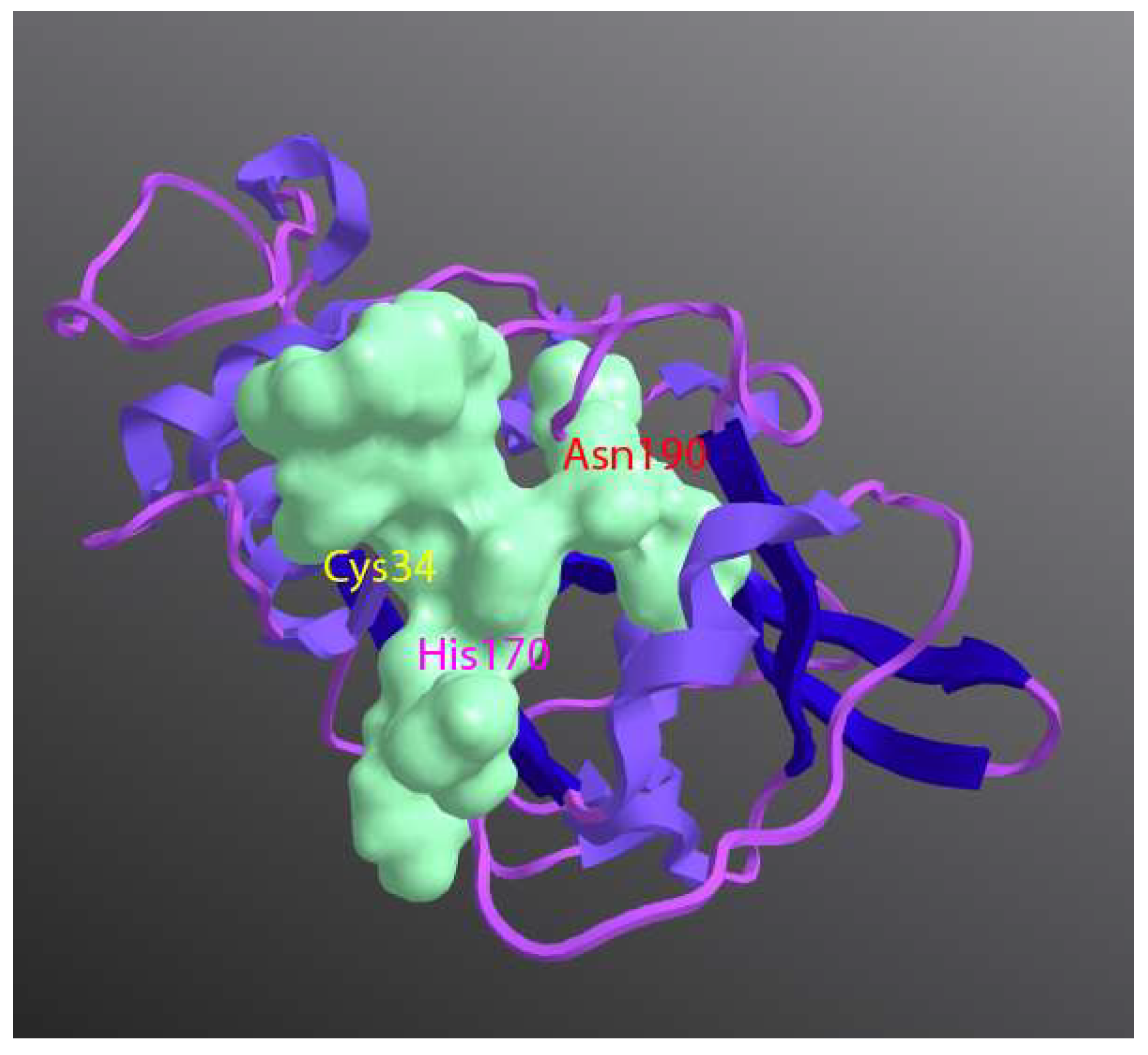{kind=link}
{kind=link}
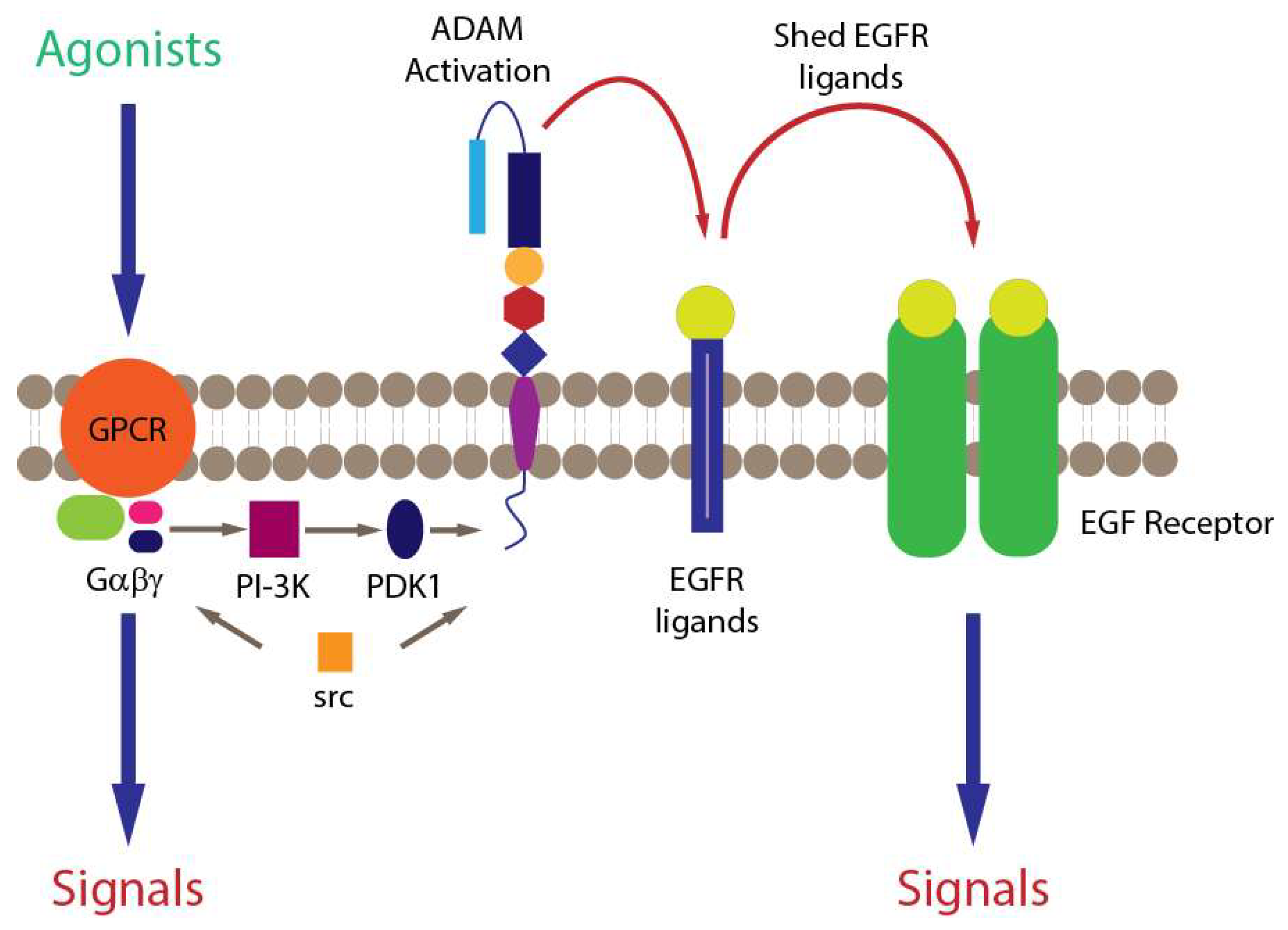{kind=link}
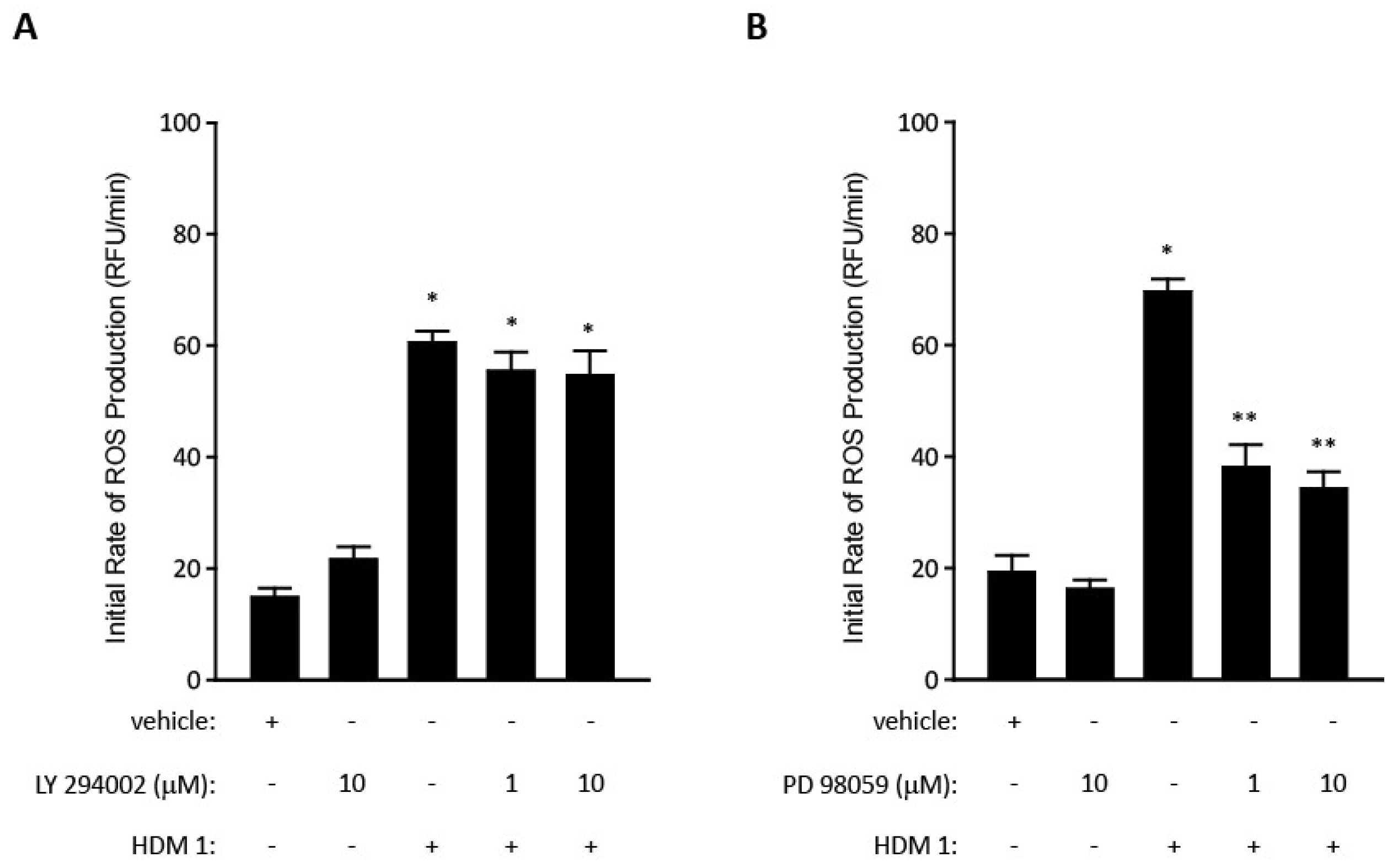{kind=link}
{kind=link}
{kind=link}
| Der p 1 | Der f 1 | Eur m 1 | cat B | cat K | cat L2 | cat S | cat Z | |
|---|---|---|---|---|---|---|---|---|
| Der p 1 | 100 | 82 | 85 | 25 | 34 | 31 | 31 | 25 |
| Der f 1 | 82 | 100 | 85 | 26 | 34 | 33 | 30 | 27 |
| Eur m 1 | 85 | 85 | 100 | 25 | 34 | 32 | 30 | 25 |
| cat B | 25 | 26 | 25 | 100 | 28 | 30 | 33 | 24 |
| cat K | 34 | 34 | 34 | 28 | 100 | 59 | 58 | 28 |
| cat L2 | 31 | 33 | 32 | 30 | 59 | 100 | 58 | 30 |
| cat S | 31 | 30 | 30 | 33 | 58 | 58 | 100 | 31 |
| cat Z | 25 | 27 | 25 | 24 | 28 | 30 | 31 | 100 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Chen, J.; Robinson, C. Cellular and Molecular Events in the Airway Epithelium Defining the Interaction Between House Dust Mite Group 1 Allergens and Innate Defences. Int. J. Mol. Sci. 2018, 19, 3549. https://doi.org/10.3390/ijms19113549
Zhang J, Chen J, Robinson C. Cellular and Molecular Events in the Airway Epithelium Defining the Interaction Between House Dust Mite Group 1 Allergens and Innate Defences. International Journal of Molecular Sciences. 2018; 19(11):3549. https://doi.org/10.3390/ijms19113549
Chicago/Turabian StyleZhang, Jihui, Jie Chen, and Clive Robinson. 2018. "Cellular and Molecular Events in the Airway Epithelium Defining the Interaction Between House Dust Mite Group 1 Allergens and Innate Defences" International Journal of Molecular Sciences 19, no. 11: 3549. https://doi.org/10.3390/ijms19113549
APA StyleZhang, J., Chen, J., & Robinson, C. (2018). Cellular and Molecular Events in the Airway Epithelium Defining the Interaction Between House Dust Mite Group 1 Allergens and Innate Defences. International Journal of Molecular Sciences, 19(11), 3549. https://doi.org/10.3390/ijms19113549