Effects of Cardiovascular Risk Factors on Cardiac STAT3



Abstract
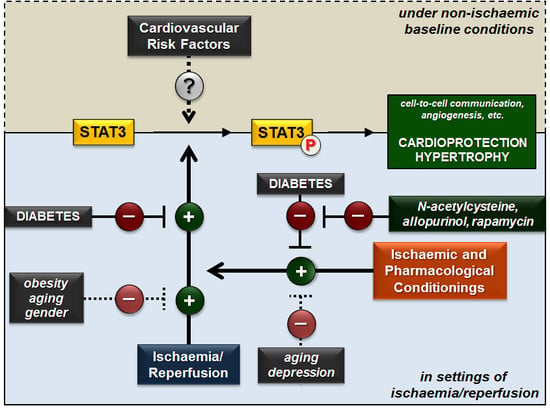
:
1. Introduction
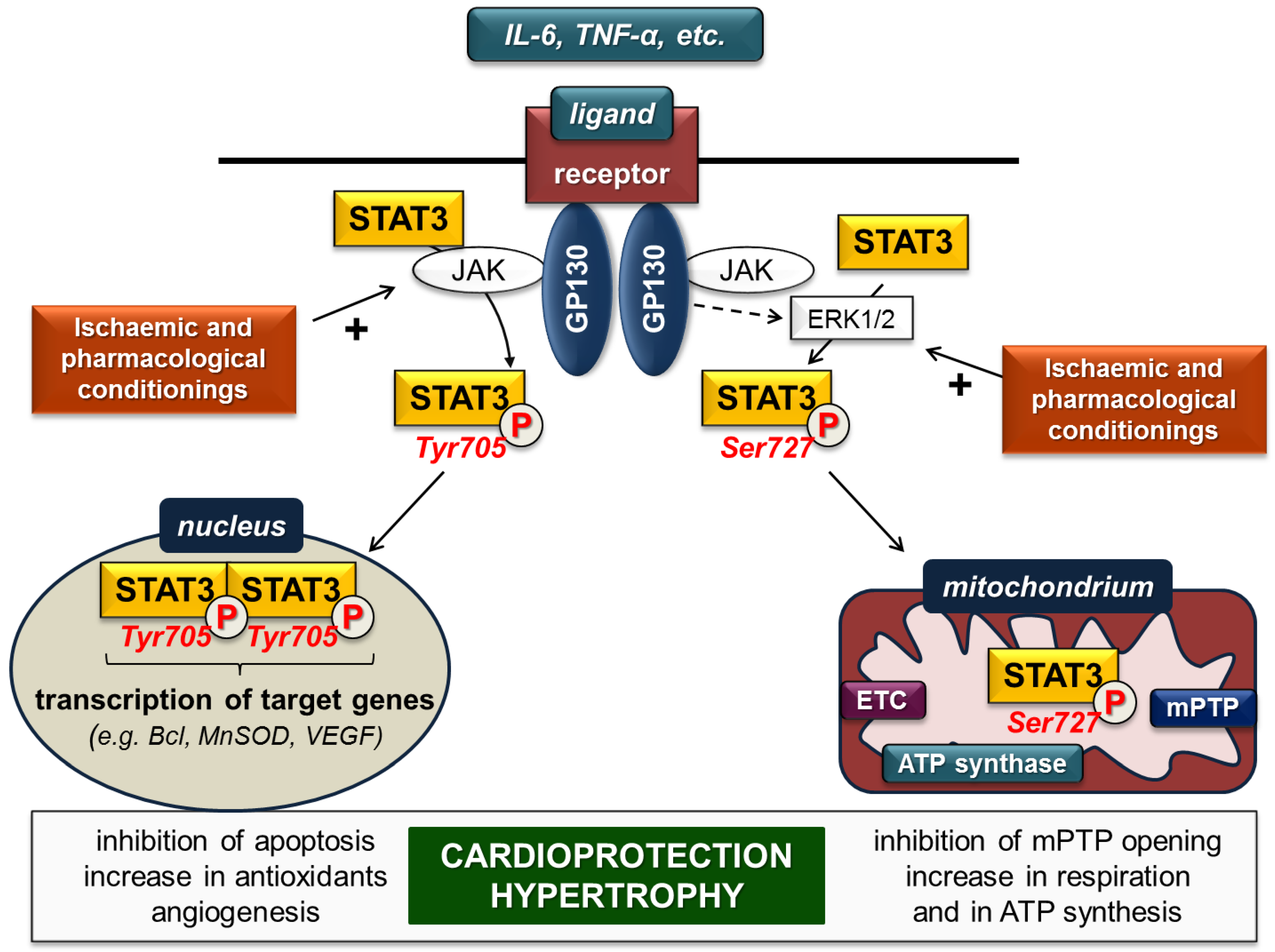
1.1. Structure of STAT3
1.2. Signalling
1.3. STAT3 in the Heart
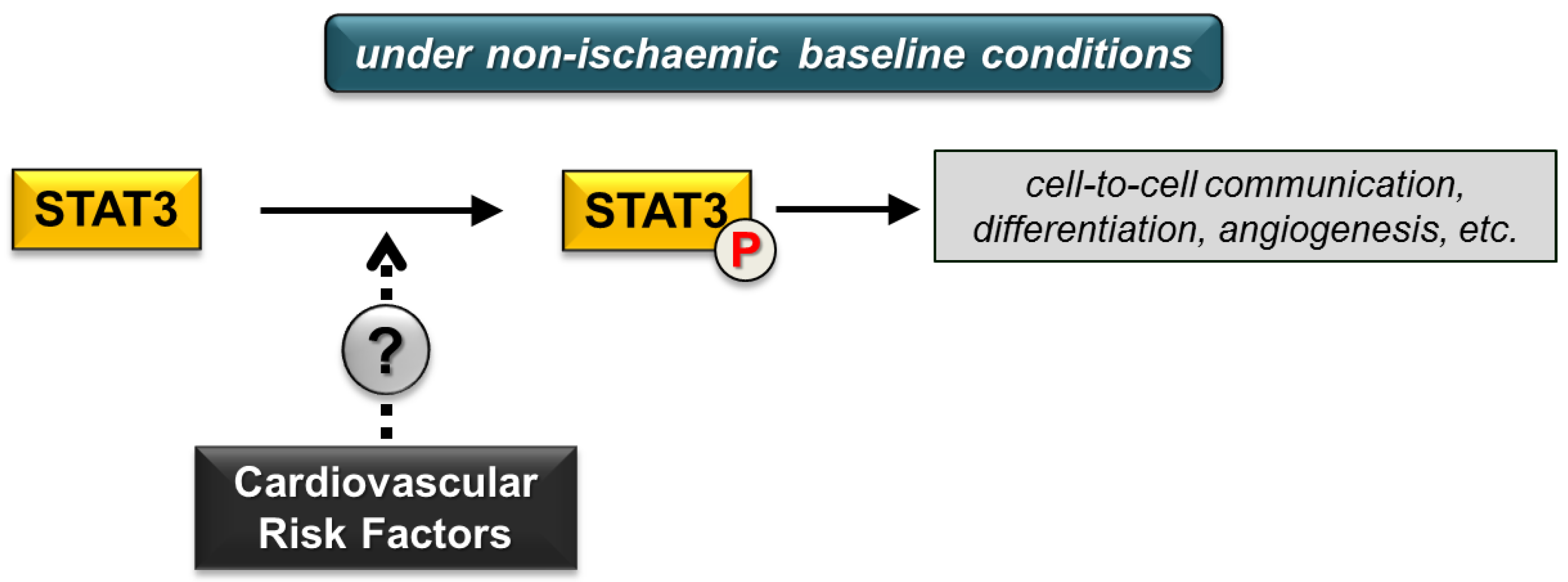
2. Effect of Cardiovascular Risk Factors on Cardiac STAT3 under Non-Ischaemic Baseline Conditions
2.1. Diabetes
2.2. Obesity
2.3. Hypertension
2.4. Chronic Kidney Disease
2.5. Aging
2.6. Smoking
2.7. Alcohol
2.8. Comedications
2.9. Summary
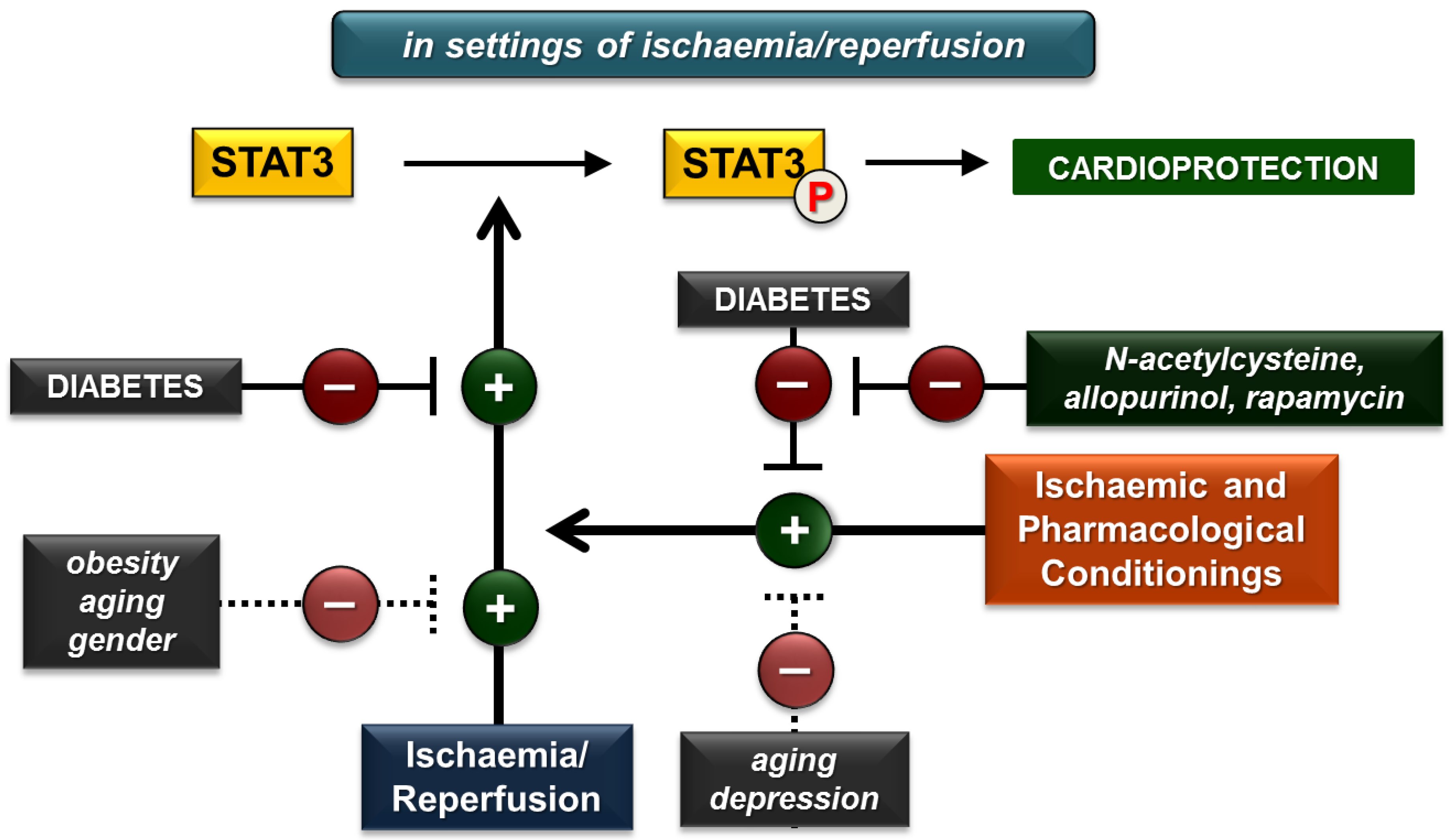
3. Effect of Cardiovascular Risk Factors on Cardiac STAT3 Activation in Settings of Ischaemia/Reperfusion
3.1. Diabetes
3.2. Obesity
3.3. Chronic Kidney Disease
3.4. Aging
3.5. Gender
3.6. Depression
3.7. Comedications
4. Effect of Cardioprotective Strategies against Ischaemia/Reperfusion on Cardiac STAT3 Activation in the Presence of Cardiovascular Risk Factors
4.1. Diabetes
4.2. Obesity
4.3. Chronic Kidney Disease
4.4. Aging
4.5. Depression
4.6. Comedications
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Bharadwaj, U.; Kasembeli, M.M.; Eckols, T.K.; Kolosov, M.; Lang, P.; Christensen, K.; Edwards, D.P.; Tweardy, D.J. Monoclonal antibodies specific for STAT3β reveal its contribution to constitutive STAT3 phosphorylation in breast cancer. Cancers 2014, 6, 2012–2034. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.; Hilfiker-Kleiner, D. Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br. J. Pharmacol. 2008, 153, S414–S427. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Rincon, M. Mitochondrial STAT3, the need for design thinking. Int. J. Biol. Sci. 2016, 12, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Garama, D.J.; White, C.L.; Balic, J.J.; Gough, D.J. Mitochondrial STAT3: Powering up a potent factor. Cytokine 2016, 87, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghikia, A.; Ricke-Hoch, M.; Stapel, B.; Gorst, I.; Hilfiker-Kleiner, D. STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc. Res. 2014, 102, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, M.; Huang, X.Y.; Zhang, J.J. STAT3 directly controls the expression of TBX5, NKX2.5, and GATA4 and is essential for cardiomyocyte differentiation of P19Cl6 cells. J. Biol. Chem. 2010, 285, 23639–23646. [Google Scholar] [CrossRef] [PubMed]
- Osugi, T.; Oshima, Y.; Fujio, Y.; Funamoto, M.; Yamashita, A.; Negoro, S.; Kunisada, K.; Izumi, M.; Nakaoka, Y.; Hirota, H.; et al. Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J. Biol. Chem. 2002, 277, 6676–6681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qu, X.; Chen, B.; Snyder, M.; Wang, M.; Li, B.; Tang, Y.; Chen, H.; Zhu, W.; Zhan, L.; et al. Critical roles of STAT3 in beta-adrenergic functions in the heart. Circulation 2016, 133, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Fuchs, M.; Kaminski, K.; Schaefer, A.; Schieffer, B.; Hillmer, A.; Schmiedl, A.; Ding, Z.; Podewski, E.; et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004, 95, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Drexler, H. Many good reasons to have STAT3 in the heart. Pharmacol. Therap. 2005, 107, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Stapel, B.; Hoch, M.; Hilfiker-Kleiner, D. STAT3 and cardiac remodeling. Heart Fail. Rev. 2011, 16, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Fujio, Y.; Nakanishi, T.; Itoh, N.; Yamamoto, Y.; Negoro, S.; Tanaka, K.; Kishimoto, T.; Kawase, I.; Azuma, J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc. Res. 2005, 65, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negoro, S.; Kunisada, K.; Tone, E.; Funamoto, M.; Oh, H.; Kishimoto, T.; Yamauchi-Takihara, K. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc. Res. 2000, 47, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, M.; Hilfiker, A.; Kaminski, K.; Hilfiker-Kleiner, D.; Guener, Z.; Klein, G.; Schieffer, B.; Rose-John, S.; Drexler, H. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. FASEB J. 2003, 17, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Zouein, F.A.; Altara, R.; Chen, Q.; Lesnefsky, E.J.; Kurdi, M.; Booz, G.W. Pivotal importance of STAT3 in protecting the heart from acute and chronic stress: New advancement and unresolved issues. Front. Cardiovasc. Med. 2015, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.-T.; Guo, Y.; Han, H.; Zhu, Y.; Bolli, R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2001, 98, 9050. [Google Scholar] [CrossRef] [PubMed]
- Skyschally, A.; Kleinbongard, P.; Lieder, H.; Gedik, N.; Stoian, L.; Amanakis, G.; Elbers, E.; Heusch, G. Humoral transfer and intramyocardial signal transduction of protection by remote ischemic perconditioning in pigs, rats, and mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H159–H172. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Buechert, A.; Heinen, Y.; Roeskes, C.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ. Res. 2008, 102, 131–135. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, K.E.; Breen, E.P.; Gallagher, H.C.; Buggy, D.J.; Hurley, J.P. Understanding STAT3 signaling in cardiac ischemia. Basic Res. Cardiol. 2016, 111, 27. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142. [Google Scholar] [CrossRef] [PubMed]
- Federation, I.D. IDF Diabetes Atlas, 8th Edition. Online Publication of International Diabetes Federation, 2017. Available online: http://www.diabetesatlas.org/.
- Otto, S.; Seeber, M.; Fujita, B.; Kretzschmar, D.; Ferrari, M.; Goebel, B.; Figulla, H.R.; Poerner, T.C. Microembolization and myonecrosis during elective percutaneous coronary interventions in diabetic patients: An intracoronary doppler ultrasound study with 2-year clinical follow-up. Basic Res. Cardiol. 2012, 107, 289. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Rahman, T.; Ismail, A.A.; Rashid, A.R. Diabetes-associated macrovasculopathy: Pathophysiology and pathogenesis. Diabetes Obes. Metab. 2007, 9, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, M.; Szucs, G.; Fekete, V.; Pipicz, M.; Eder, K.; Gaspar, R.; Soja, A.; Pipis, J.; Ferdinandy, P.; Csonka, C.; et al. Transcriptomic alterations in the heart of non-obese type 2 diabetic goto-kakizaki rats. Cardiovasc. Diabetol. 2016, 15, 110. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, M.; Szucs, G.; Pipicz, M.; Zvara, A.; Eder, K.; Fekete, V.; Szucs, C.; Barkanyi, J.; Csonka, C.; Puskas, L.G.; et al. The effect of a preparation of minerals, vitamins and trace elements on the cardiac gene expression pattern in male diabetic rats. Cardiovasc. Diabetol. 2015, 14, 85. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, M.; Zvara, A.; Gyemant, N.; Fekete, V.; Kocsis, G.F.; Pipis, J.; Szucs, G.; Csonka, C.; Puskas, L.G.; Ferdinandy, P.; et al. Metabolic syndrome influences cardiac gene expression pattern at the transcript level in male zdf rats. Cardiovasc. Diabetol. 2013, 12, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Qiao, S.; Lei, S.; Liu, Y.; Ng, K.F.; Xu, A.; Lam, K.S.; Irwin, M.G.; Xia, Z. N-acetylcysteine and allopurinol synergistically enhance cardiac adiponectin content and reduce myocardial reperfusion injury in diabetic rats. PLoS ONE 2011, 6, e23967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Li, H.; Wang, S.; Mao, X.; Yan, D.; Wong, S.S.; Xia, Z.; Irwin, M.G. Repeated non-invasive limb ischemic preconditioning confers cardioprotection through PKC-/STAT3 signaling in diabetic rats. Cell Physiol. Biochem. 2018, 45, 2107–2121. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lei, S.; Liu, Y.; Gao, X.; Irwin, M.G.; Xia, Z.Y.; Hei, Z.; Gan, X.; Wang, T.; Xia, Z. Antioxidant N-acetylcysteine attenuates the reduction of BRG1 protein expression in the myocardium of type 1 diabetic rats. J. Diabetes Res. 2013, 2013, 716219. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Lei, S.; Xia, Z.Y.; Wu, Y.; Meng, Q.; Zhan, L.; Su, W.; Liu, H.; Xu, J.; Liu, Z.; et al. Selective inhibition of pten preserves ischaemic post-conditioning cardioprotection in stz-induced type 1 diabetic rats: Role of the PI3k/AKT and JAK2/STAT3 pathways. Clin. Sci. 2016, 130, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Wang, S.; Zhang, L.; Xie, X.; Cai, S.; Li, H.; Xie, G.L.; Miao, H.L.; Yang, C.; Liu, X.; et al. Propofol through upregulating caveolin-3 attenuates post-hypoxic mitochondrial damage and cell death in H9C2 cardiomyocytes during hyperglycemia. Cell Physiol. Biochem. 2017, 44, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Huang, H.; Liu, S.; Mao, X.; Wang, S.; Wong, S.S.; Xia, Z.; Irwin, M.G. Cardioprotection from emulsified isoflurane postconditioning is lost in rats with streptozotocin-induced diabetes due to the impairment of BRG1/NRF2/STAT3 signalling. Clin. Sci. 2016, 130, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, R.C.; Yang, Z.H.; Sun, G.B.; Wang, M.; Ma, X.J.; Yang, L.J.; Sun, X.B. Taxifolin prevents diabetic cardiomyopathy in vivo and in vitro by inhibition of oxidative stress and cell apoptosis. Food Chem. Toxicol. 2014, 63, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Li, D. Losartan reduces myocardial interstitial fibrosis in diabetic cardiomyopathy rats by inhibiting JAK/STAT signaling pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 466–473. [Google Scholar] [PubMed]
- Lo, S.H.; Hsu, C.T.; Niu, H.S.; Niu, C.S.; Cheng, J.T.; Chen, Z.C. Ginsenoside RH2 improves cardiac fibrosis via PPARδ-STAT3 signaling in type 1-like diabetic rats. Int. J. Mol. Sci. 2017, 18, 1364. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.T.; Cheng, J.T.; Chen, Z.C. Telmisartan improves cardiac fibrosis in diabetes through peroxisome proliferator activated receptor delta (PPARδ): From bedside to bench. Cardiovasc. Diabetol. 2016, 15, 113. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Cui, M.; Zhu, M.; Su, W.L.; Qiu, M.C.; Zhang, H. STAT1/3 and ERK1/2 synergistically regulate cardiac fibrosis induced by high glucose. Cell Physiol. Biochem. 2013, 32, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.O.; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Gadde, K.M.; Martin, C.K.; Berthoud, H.R.; Heymsfield, S.B. Obesity: Pathophysiology and management. J. Am. Coll. Cardiol. 2018, 71, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M. Cardiac adiposity and global cardiometabolic risk: New concept and clinical implication. Circ. J. 2009, 73, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N.; Kok, B.P.; Kienesberger, P.C.; Lehner, R.; Dyck, J.R. Shedding light on the enigma of myocardial lipotoxicity: The involvement of known and putative regulators of fatty acid storage and mobilization. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E897–E908. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Perego, L.; Pizzocri, P.; Corradi, D.; Maisano, F.; Paganelli, M.; Fiorina, P.; Barbieri, M.; Morabito, A.; Paolisso, G.; Folli, F.; et al. Circulating leptin correlates with left ventricular mass in morbid (grade III) obesity before and after weight loss induced by bariatric surgery: A potential role for leptin in mediating human left ventricular hypertrophy. J. Clin. Endocrinol. Metab. 2005, 90, 4087–4093. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Tagliamonte, M.R.; Galderisi, M.; Zito, G.A.; Petrocelli, A.; Carella, C.; de Divitiis, O.; Varricchio, M. Plasma leptin level is associated with myocardial wall thickness in hypertensive insulin-resistant men. Hypertension 1999, 34, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Madani, S.; De Girolamo, S.; Munoz, D.M.; Li, R.K.; Sweeney, G. Direct effects of leptin on size and extracellular matrix components of human pediatric ventricular myocytes. Cardiovasc. Res. 2006, 69, 716–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajapurohitam, V.; Gan, X.T.; Kirshenbaum, L.A.; Karmazyn, M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ. Res. 2003, 93, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Rajapurohitam, V.; Javadov, S.; Purdham, D.M.; Kirshenbaum, L.A.; Karmazyn, M. An autocrine role for leptin in mediating the cardiomyocyte hypertrophic effects of angiotensin II and endothelin-1. J. Mol. Cell Cardiol. 2006, 41, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Kloek, C.; Haq, A.K.; Dunn, S.L.; Lavery, H.J.; Banks, A.S.; Myers, M.G., Jr. Regulation of JAK kinases by intracellular leptin receptor sequences. J. Biol. Chem. 2002, 277, 41547–41555. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.T.; Zhao, G.; Huang, C.X.; Rowe, A.C.; Purdham, D.M.; Karmazyn, M. Identification of fat mass and obesity associated (FTO) protein expression in cardiomyocytes: Regulation by leptin and its contribution to leptin-induced hypertrophy. PLoS ONE 2013, 8, e74235. [Google Scholar] [CrossRef] [PubMed]
- Leifheit-Nestler, M.; Wagner, N.M.; Gogiraju, R.; Didie, M.; Konstantinides, S.; Hasenfuss, G.; Schafer, K. Importance of leptin signaling and signal transducer and activator of transcription-3 activation in mediating the cardiac hypertrophy associated with obesity. J. Transl. Med. 2013, 11, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.K.; Yeh, Y.L.; Lin, Y.M.; Lin, J.Y.; Tzang, B.S.; Lin, J.A.; Yang, A.L.; Wu, F.L.; Tsai, F.J.; Cheng, S.M.; et al. Cardiac hypertrophy-related pathways in obesity. Chin. J. Physiol. 2014, 57, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Wu, C.H.; Kuo, W.W.; Lin, J.A.; Wang, H.F.; Tsai, F.J.; Tsai, C.H.; Huang, C.Y.; Hsu, T.C.; Tzang, B.S. Ameliorate effects of Li-Fu formula on IL-6-mediated cardiac hypertrophy in hamsters fed with a hyper-cholesterol diet. Evid. Based Complement. Altern. Med. 2011, 2011, 485471. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhang, Y.; Xin, L.; Kong, S.; Chen, Y.; Yang, S.; Li, K. Global transcriptomic profiling of cardiac hypertrophy and fatty heart induced by long-term high-energy diet in bama miniature pigs. PLoS ONE 2015, 10, e0132420. [Google Scholar] [CrossRef] [PubMed]
- Phan, W.L.; Huang, Y.T.; Ma, M.C. Interleukin-27 protects cardiomyocyte-like H9C2 cells against metabolic syndrome: Role of STAT3 signaling. BioMed Res. Int. 2015, 2015, 689614. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.S.; Donthamsetty, S.; White, B.; Mehendale, H.M. High fat diet-fed obese rats are highly sensitive to doxorubicin-induced cardiotoxicity. Toxicol. Appl. Pharmacol. 2008, 231, 413–422. [Google Scholar] [CrossRef] [PubMed]
- McGaffin, K.R.; Sun, C.K.; Rager, J.J.; Romano, L.C.; Zou, B.; Mathier, M.A.; O’Doherty, R.M.; McTiernan, C.F.; O’Donnell, C.P. Leptin signalling reduces the severity of cardiac dysfunction and remodelling after chronic ischaemic injury. Cardiovasc. Res. 2008, 77, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.V.; Zheng, M.; Schuleri, K.H.; Phan, A.C.; Bedja, D.; Saraiva, R.M.; Yiginer, O.; Vandegaer, K.; Gabrielson, K.L.; O’Donnell, C.P.; et al. Activation of the cardiac ciliary neurotrophic factor receptor reverses left ventricular hypertrophy in leptin-deficient and leptin-resistant obesity. Proc. Natl. Acad. Sci. USA 2006, 103, 4222–4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APHA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. Hypertension 2018, 71, e13–e115. [Google Scholar] [PubMed]
- Lewington, S.; Clarke, R.; Qizilbash, N.; Peto, R.; Collins, R.; Prospective Studies, C. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002, 360, 1903–1913. [Google Scholar] [PubMed]
- Rapsomaniki, E.; Timmis, A.; George, J.; Pujades-Rodriguez, M.; Shah, A.D.; Denaxas, S.; White, I.R.; Caulfield, M.J.; Deanfield, J.E.; Smeeth, L.; et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1.25 million people. Lancet 2014, 383, 1899–1911. [Google Scholar] [CrossRef]
- Egan, B.M.; Li, J.; Hutchison, F.N.; Ferdinand, K.C. Hypertension in the united states, 1999 to 2012: Progress toward healthy people 2020 goals. Circulation 2014, 130, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Blood Pressure Lowering Treatment Trialists, C. Blood pressure-lowering treatment based on cardiovascular risk: A meta-analysis of individual patient data. Lancet 2014, 384, 591–598. [Google Scholar]
- Pan, J.; Fukuda, K.; Kodama, H.; Makino, S.; Takahashi, T.; Sano, M.; Hori, S.; Ogawa, S. Role of angiotensin ii in activation of the JAK/STAT pathway induced by acute pressure overload in the rat heart. Circ. Res. 1997, 81, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Booz, G.W.; Day, J.N.; Baker, K.M. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: Role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J. Mol. Cell Cardiol. 2002, 34, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.; Hilfiker-Kleiner, D. Survival pathways in hypertrophy and heart failure: The GP130-STAT axis. Basic Res. Cardiol. 2007, 102, 393–411. [Google Scholar] [CrossRef] [PubMed]
- Wincewicz, A.; Sulkowski, S. STAT proteins as intracellular regulators of resistance to myocardial injury in the context of cardiac remodeling and targeting for therapy. Adv. Clin. Exp. Med. 2017, 26, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Matsushita, K.; Abate, K.H.; Al-Aly, Z.; Arnlov, J.; Asayama, K.; Atkins, R.; Badawi, A.; Ballew, S.H.; Banerjee, A.; et al. Global cardiovascular and renal outcomes of reduced GFR. J. Am. Soc. Nephrol. 2017, 28, 2167–2179. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Hsieh, C.L.; Peng, C.C.; Peng, R.Y. Exercise rescued chronic kidney disease by attenuating cardiac hypertrophy through the cardiotrophin-1 -> LIFR/gp 130 -> jak/STAT3 pathway. Eur. J. Prev. Cardiol. 2014, 21, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.P.; Hsieh, C.H.; Ho, T.J.; Kuo, W.W.; Yeh, Y.L.; Lin, C.C.; Kuo, C.H.; Huang, C.Y. Secondhand smoke exposure toxicity accelerates age-related cardiac disease in old hamsters. BMC Cardiovasc. Disord. 2014, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.P.; Hsieh, D.J.; Kuo, W.W.; Han, C.K.; Pai, P.; Yeh, Y.L.; Lin, C.C.; Padma, V.V.; Day, C.H.; Huang, C.Y. Secondhand smoke exposure reduced the compensatory effects of IGF-I growth signaling in the aging rat hearts. Int. J. Med. Sci. 2015, 12, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Ren, J. Mtor-STAT3-notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. J. Cell Mol. Med. 2012, 16, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Sander, M.; Oxlund, B.; Jespersen, A.; Krasnik, A.; Mortensen, E.L.; Westendorp, R.G.J.; Rasmussen, L.J. The challenges of human population ageing. Age Ageing 2015, 44, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Castelli, W.P. Epidemiology of coronary heart disease: The framingham study. Am. J. Med. 1984, 76, 4–12. [Google Scholar] [CrossRef]
- Jousilahti, P.; Vartiainen, E.; Tuomilehto, J.; Puska, P. Sex, age, cardiovascular risk factors, and coronary heart disease: A prospective follow-up study of 14 786 middle-aged men and women in Finland. Circulation 1999, 99, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.; Sundström, J.; Ärnlöv, J.; Lampa, E. Impact of aging on the strength of cardiovascular risk factors: A longitudinal study over 40 years. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, J.J.; Kalinowski, A.; Liu, M.G.; Zhang, S.S.; Gao, Q.; Chai, G.X.; Ji, L.; Iwamoto, Y.; Li, E.; Schneider, M.; et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc. Natl. Acad. Sci. USA 2003, 100, 12929–12934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castello, L.; Maina, M.; Testa, G.; Cavallini, G.; Biasi, F.; Donati, A.; Leonarduzzi, G.; Bergamini, E.; Poli, G.; Chiarpotto, E. Alternate-day fasting reverses the age-associated hypertrophy phenotype in rat heart by influencing the ERK and PI3K signaling pathways. Mech. Ageing Dev. 2011, 132, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Quan, J.; Johnston, W.E.; Maass, D.L.; Horton, J.W.; Thomas, J.A.; Tao, W. Age-dependent differences of interleukin-6 activity in cardiac function after burn complicated by sepsis. Burns J. Int. Soc. Burn Inj. 2010, 36, 232–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Chen, D.; Fang, N.; Yao, Y.; Li, L. Age-associated differences in response to sevoflurane postconditioning in rats. Scand. Cardiovasc. J. 2016, 50, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Jiang, J.; Geng, Y.J. Attenuated expression of gelsolin in association with induction of aquaporin-1 and nitric oxide synthase in dysfunctional hearts of aging mice exposed to endotoxin. Int. J. Immunopathol. Pharmacol. 2012, 25, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Harries, L.W.; Fellows, A.D.; Pilling, L.C.; Hernandez, D.; Singleton, A.; Bandinelli, S.; Guralnik, J.; Powell, J.; Ferruci, L.; Melzer, D. Advancing age is associated with gene expression changes resembling mtor inhibition: Evidence from two human populations. Mech. Ageing Dev. 2012, 133, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.R.; Woodward, M. Cigarette smoking as a risk factor for coronary heart disease in women compared with men: A systematic review and meta-analysis of prospective cohort studies. Lancet 2011, 378, 1297–1305. [Google Scholar] [CrossRef]
- Zhao, J.; Stockwell, T.; Roemer, A.; Naimi, T.; Chikritzhs, T. Alcohol consumption and mortality from coronary heart disease: An updated meta-analysis of cohort studies. J. Stud. Alcohol. Drugs 2017, 78, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Rajtik, T.; Carnicka, S.; Szobi, A.; Mesarosova, L.; Matus, M.; Svec, P.; Ravingerova, T.; Adameova, A. Pleiotropic effects of simvastatin are associated with mitigation of apoptotic component of cell death upon lethal myocardial reperfusion-induced injury. Physiol. Res. 2012, 61, S33–S41. [Google Scholar] [PubMed]
- Al-rasheed, N.M.; Al-Oteibi, M.M.; Al-Manee, R.Z.; Al-shareef, S.A.; Al-Rasheed, N.M.; Hasan, I.H.; Mohamad, R.A.; Mahmoud, A.M. Simvastatin prevents isoproterenol-induced cardiac hypertrophy through modulation of the JAK/STAT pathway. Drug Des. Dev. Ther. 2015, 9, 3217–3229. [Google Scholar]
- Li, H.; Yao, W.; Liu, Z.; Xu, A.; Huang, Y.; Ma, X.L.; Irwin, M.G.; Xia, Z. Hyperglycemia abrogates ischemic postconditioning cardioprotection by impairing AdiPor1/caveolin-3/STAT3 signaling in diabetic rats. Diabetes 2016, 65, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Itoh, T.; Sunaga, D.; Miura, T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc. Diabetol. 2012, 11, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Mao, X.; Li, H.; Qiao, S.; Xu, A.; Wang, J.; Lei, S.; Liu, Z.; Ng, K.F.; Wong, G.T.; et al. N-acetylcysteine and allopurinol up-regulated the JAK/STAT3 and PI3K/AKT pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic. Biol. Med. 2013, 63, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Diabetes abolishes morphine-induced cardioprotection via multiple pathways upstream of glycogen synthase kinase-3β. Diabetes 2007, 56, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wang, T.; Li, Y.; Wang, M.; Li, H.; Irwin, M.G.; Xia, Z. N-acetylcysteine restores sevoflurane postconditioning cardioprotection against myocardial ischemia-reperfusion injury in diabetic rats. J. Diabetes Res. 2016, 2016, 9213034. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Salloum, F.N.; Filippone, S.M.; Durrant, D.E.; Rokosh, G.; Bolli, R.; Kukreja, R.C. Inhibition of mammalian target of rapamycin protects against reperfusion injury in diabetic heart through STAT3 signaling. Basic Res. Cardiol. 2015, 110, 31. [Google Scholar] [CrossRef] [PubMed]
- McGaffin, K.R.; Witham, W.G.; Yester, K.A.; Romano, L.C.; O’Doherty, R.M.; McTiernan, C.F.; O’Donnell, C.P. Cardiac-specific leptin receptor deletion exacerbates ischaemic heart failure in mice. Cardiovasc. Res. 2011, 89, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Mocanu, M.M.; Davidson, S.M.; Wynne, A.M.; Simpkin, J.C.; Yellon, D.M. Leptin, the obesity-associated hormone, exhibits direct cardioprotective effects. Br. J. Pharmacol. 2006, 149, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadou, I.; Benaki, D.; Efentakis, P.; Bibli, S.I.; Milioni, A.I.; Papachristodoulou, A.; Zoga, A.; Skaltsounis, A.L.; Mikros, E.; Iliodromitis, E.K. The natural olive constituent oleuropein induces nutritional cardioprotection in normal and cholesterol-fed rabbits: Comparison with preconditioning. Planta Med. 2015, 81, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.J.; McCafferty, K.; Kieswich, J.; Harwood, S.; Andrikopoulos, P.; Raftery, M.; Thiemermann, C.; Yaqoob, M.M. Ischemic conditioning protects the uremic heart in a rodent model of myocardial infarction. Circulation 2012, 125, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, P.; Chen, M.; Zhang, W.; Yu, L.; Yang, X.C.; Fan, Q. Aging might increase myocardial ischemia/reperfusion-induced apoptosis in humans and rats. Age 2012, 34, 621–632. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Toyoda, Y.; Wakiyama, H.; Rousou, A.J.; Parker, R.A.; Levitsky, S. Age- and gender-related differences in ischemia/reperfusion injury and cardioprotection: Effects of diazoxide. Ann. Thorac. Surg. 2006, 82, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Azhar, G.; Gao, W.; Liu, L.; Wei, J.Y. Ischemia-reperfusion in the adult mouse heart influence of age. Exp. Gerontol. 1999, 34, 699–714. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Gallo, D.S.; Ye, J.; Whittingham, T.S.; Lust, W.D. Aging increases ischemia-reperfusion injury in the isolated, buffer-perfused heart. J. Lab. Clin. Med. 1994, 124, 843–851. [Google Scholar] [PubMed]
- Mendelsohn, M.E.; Karas, R.H. Molecular and cellular basis of cardiovascular gender differences. Science 2005, 308, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, M.T. Mechanisms of cardioprotection by estrogens. Proc. Soc. Exp. Biol. Med. 1998, 217, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Crisostomo, P.R.; Markel, T.A.; Wang, Y.; Meldrum, D.R. Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation 2008, 118, S38–S45. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Y.; Abarbanell, A.; Tan, J.; Weil, B.; Herrmann, J.; Meldrum, D.R. Both endogenous and exogenous testosterone decrease myocardial STAT3 activation and SOCS3 expression after acute ischemia and reperfusion. Surgery 2009, 146, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, J.H.; Bigger, J.T.; Blumenthal, J.A.; Frasure-Smith, N.; Kaufmann, P.G.; Lespérance, F.; Mark, D.B.; Sheps, D.S.; Taylor, C.B.; Froelicher, E.S. Depression and coronary heart disease. Endor. Am. Psychiatr. Assoc. 2008, 118, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Wang, Y.; Wang, X.; Wang, Y.; Chen, Y. Cardioprotection by ischemic postconditioning is abolished in depressed rats: Role of AKT and signal transducer and activator of transcription-3. Mol. Cell Biochem. 2011, 346, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Maynard, M.; Greiner, D.L.; Whittaker, P. Cardioprotection with postconditioning: Loss of efficacy in murine models of type-2 and type-1 diabetes. Antioxid. Redox Signal. 2011, 14, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Tsang, A.; Hausenloy, D.J.; Mocanu, M.M.; Carr, R.D.; Yellon, D.M. Preconditioning the diabetic heart: The importance of Akt phosphorylation. Diabetes 2005, 54, 2360–2364. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Cho, S.; Tosaka, S.; Ureshino, H.; Maekawa, T.; Hara, T.; Sumikawa, K. Pharmacological preconditioning in type 2 diabetic rat hearts: The roles of mitochondrial ATP-sensitive potassium channels and the phosphatidylinositol 3-kinase-Akt pathway. Cardiovasc. Drugs Ther. 2009, 23, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, J.A.; Lofgren, B.; Rasmussen, L.E.; Nielsen, J.M.; Norregaard, R.; Kristiansen, S.B.; Botker, H.E.; Nielsen, T.T. Cardioprotective effect of l-glutamate in obese type 2 diabetic zucker fatty rats. Clin. Exp. Pharmacol. Physiol. 2009, 36, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Samidurai, A.; Salloum, F.N.; Durrant, D.; Chernova, O.B.; Kukreja, R.C.; Das, A. Chronic treatment with novel nanoformulated micelles of rapamycin, rapatar, protects diabetic heart against ischaemia/reperfusion injury. Br. J. Pharmacol. 2017, 174, 4771–4784. [Google Scholar] [CrossRef] [PubMed]
- Csont, T.; Ferdinandy, P. Letter by csont and ferdinandy regarding article, ischemic conditioning protects the uremic heart in a rodent model of myocardial infarction. Circulation 2012, 126, e212; author reply e213. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, G.F.; Sarkozy, M.; Bencsik, P.; Pipicz, M.; Varga, Z.V.; Paloczi, J.; Csonka, C.; Ferdinandy, P.; Csont, T. Preconditioning protects the heart in a prolonged uremic condition. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1229–H1236. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, G.; Abete, P.; Ferrara, N.; Papa, A.; Rosiello, R.; Furgi, G.; Calabrese, C.; Cacciatore, F.; Rengo, F. Warm-up phenomenon in adult and elderly patients with coronary artery disease: Further evidence of the loss of ischemic preconditioning in the aging heart. J. Gerontol. A Biol. Sci. Med. Sci. 2000, 55, M124–M129. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-K.; Pehkonen, E.; Laurikka, J.; Kaukinen, L.; Honkonen, E.L.; Kaukinen, S.; Laippala, P.; Tarkka, M.R. The protective effects of preconditioning decline in aged patients undergoing coronary artery bypass grafting. J. Thor. Cardiovasc. Surg. 2001, 122, 972–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Konietzka, I.; Buechert, A.; Heinen, Y.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am. J. Physiol. Heart and Circul. Physiol. 2007, 292, H1764–H1769. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Schulz, R.; Heusch, G. Loss of cardioprotection with ageing. Cardiovasc. Res. 2009, 83, 247–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, E.J. Pre- and post-conditioning hormesis in elderly mice, rats, and humans: Its loss and restoration. Biogerontology 2016, 17, 681–702. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Wang, Y.; Tian, H.; Wang, X.; Chen, Y.; Mao, F. Impairment of endothelial protection by ischemic postconditioning in patients with major depressive disorder. Can. J. Physiol. Pharmacol. 2011, 89, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Csont, T. Nitroglycerin-induced preconditioning: Interaction with nitrate tolerance. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H308–H309. [Google Scholar] [CrossRef] [PubMed]
- Fekete, V.; Murlasits, Z.; Aypar, E.; Bencsik, P.; Sarkozy, M.; Szenasi, G.; Ferdinandy, P.; Csont, T. Myocardial postconditioning is lost in vascular nitrate tolerance. J. Cardiovasc. Pharmacol. 2013, 62, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, G.F.; Pipis, J.; Fekete, V.; Kovacs-Simon, A.; Odendaal, L.; Molnar, E.; Giricz, Z.; Janaky, T.; van Rooyen, J.; Csont, T.; et al. Lovastatin interferes with the infarct size-limiting effect of ischemic preconditioning and postconditioning in rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2406–H2409. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, Y.J.; Qian, H.Y.; Tang, Y.D.; Wang, H.; Zhang, Q. Rosuvastatin treatment activates JAK-STAT pathway and increases efficacy of allogeneic mesenchymal stem cell transplantation in infarcted hearts. Circ. J. 2011, 75, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Rat Sprague Dawley Male | STZ-induced diabetes | nondiabetic | ventricles | ↓ | ↓ (NC) | Ser727 | – (NC) | diabetes decreases STAT3 activation | [28] |
| LV | ↓ | ↓ | Tyr705 Ser727 | – (NC) | diabetes decreases STAT3 activation and phosphorylation | [29] | |||
| whole heart | ↓ (NC) | ↓ | Tyr705 Ser727 | – | diabetes decreases STAT3 phosphorylation | [30] | |||
| ↓ | ↓ | Tyr705 | – (NC) | diabetes decreases STAT3 activation and phosphorylation | [31] | ||||
| ↑ | ↑ | ? | ↑ | diabetes increases STAT3 activation, phosphorylation and expression | [36] | ||||
| N.D. | N.D. | N.A. | ↑ | diabetes increases STAT3 expression | [37] | ||||
| Rat prague Dawley Male | isolated adult diabetic rat cardiomyocytes | isolated adult nondiabetic rat cardiomyocytes | cells | ↓ | ↓ | Tyr705 | – (NC) | diabetes decreases STAT3 activation and phosphorylation | [33] |
| H9c2 cells | high glucose conditions | normal glucose condition | cells | ↓ | ↓ | Tyr705 | – (NC) | high glucose condition decreases STAT3 activation and phosphorylation | [32] |
| ↓ | ↓ | Tyr705 Ser727 | – (NC) | high glucose condition decreases STAT3 activation and phosphorylation | [29] | ||||
| ↑ | ↑ (NC) | ? | ↑ (NC) | high glucose condition increases STAT3 activation | [36] | ||||
| N.D. | N.D. | N.A. | ↑ | diabetes increases STAT3 expression | [37] | ||||
| Rat Wistar N.A. | isolated cardiac fibroblasts in high glucose conditions | normal glucose condition | neonatal cells | ↑ | ↑ | ? | – (NC) | high glucose condition increases STAT3 activation and phosphorylation | [38] |
| Rat Wistar Male | high-glucose and -fat diet + STZ-induced diabetes | nondiabetic | whole heart | ↑ (NC) | ↑ | ? | – (NC) | diabetes increases STAT3 phosphorylation | [35] |
| Mouse C57BL/6 Male | STZ-induced diabetes | nondiabetic | ↑ | ↑ (NC) | ? | – (NC) | diabetes increases STAT3 activation | [34] | |
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | P-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Rat Sprague Dawley Male | STZ-induced diabetes + N-acetylcysteine | STZ-induced diabetes | ventricles | ↑ | ↑ (NC) | Ser727 | – (NC) | N-acetylcysteine restores impaired activation of STAT3 in diabetes | [28] |
| whole heart | ↑ (NC) | ↑ | Tyr705 Ser727 | – | N-acetylcysteine restores impaired phosphorylation of STAT3 in diabetes | [30] | |||
| Rat Wistar Male | high-glucose and -fat diet + STZ-induced diabetes + losartan | high-glucose and -fat diet + STZ-induced diabetes | whole heart | ↓ (NC) | ↓ | ? | – (NC) | losartan attenuates enhanced phosphorylation of STAT3 in diabetes | [35] |
| Rat Sprague Dawley Male | STZ-induced diabetes + telmisartan | STZ-induced diabetes | whole heart | N.D. | N.D. | N.A. | ↓ | telmisartan attenuates enhanced expression of STAT3 in diabetes | [37] |
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Mouse C57BL/6 Unknown gender | high-fat-diet-induced obese mice | non-obese | ventricles | ↑ | ↑ (NC) | Tyr705 | – (NC) | high-fat diet increases STAT3 activation | [52] |
| leptin-receptor-deficient (db/db) obese mice | non-obese | ventricles | – | – (NC) | Tyr705 | – (NC) | STAT3 is not activated in db/db obesity | ||
| Rat Sprague-Dawley Male | high-fat-diet-induced obese rats | non-obese | whole heart | – | – (NC) | ? | – (NC) | high-fat diet did not influence STAT3 activation | [51] |
| Rat Zucker Male | leptin receptor deficient (fa/fa) obese rats | non-obese | LV | ↑ (NC) | ↑ | ? | ↑ | fa/fa genetic obesity increases STAT3 phosphorylation and expression | [53] |
| Hamster Golden Syrian Male | 0.2% cholesterol diet-induced hypercholesterolemic hamsters | normo-cholesterolemia | LV | N.D. | N.D. | N.A. | ↑ | hypercholesterolemia increases STAT3 expression | [54] |
| Pig Bama miniature Female/Male | high-fat and high-sucrose diet-induced metabolic syndrome | non-metabolic syndrome | LV | N.D. | N.D. | N.A. | ↑ * | metabolic syndrome increases STAT3 mRNA expression | [55] |
| H9c2 cells | metabolic syndrome induced by high glucose, salt, and cholesterol treatment | normal medium | cells | ↓ (ELISA) | N.E. | ? | N.E. | metabolic syndrome decreases STAT3 activation | [56] |
| Rat Sprague-Dawley Male | high-fat-diet-induced obese rats | non-obese | whole heart | ↓ (NC) | ↓ | ? | ↓ | high-fat diet decreases STAT3 phosphorylation and expression | [57] |
| Mouse Leptin-receptor-deficient (ob/ob) obese Male | leptin-receptor-deficient (ob/ob) obese mice | non-obese | whole heart | – (NC) | – | Tyr705 | – | STAT3 is not activated in ob/ob obesity | [58] |
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| HYPERTENSION | |||||||||
| Rat Wistar Male | pressure overload by abdominal aorta ligation | sham | LV | ↑ | ↑ | Tyr705 | – (NC) | pressure overload increases STAT3 activation and phosphorylation | [65] |
| CHRONIC KIDNEY DISEASE | |||||||||
| Rat Sprague-Dawley Male | doxorubicin-induced CKD sedentary | normal kidney function sedentary | whole heart | N.D. | ↑ | ? | N.D. | doxorubicin-induced CKD increases STAT3 phosphorylation | [71] |
| doxorubicin-induced CKD swimming | doxorubicin-induced CKD sedentary | whole heart | N.D. | ↓ | ? | N.D. | swimming decreases STAT3 phosphorylation in doxorubicin-induced CKD | ||
| SMOKING | |||||||||
| Hamster N.A. Male | young (6-week-old) hamsters with secondhand cigarette smoke exposure (10 cigarettes for 30 min, 4 weeks) | young hamsters without secondhand cigarette smoke exposure | LV | N.D. | N.D. | N.A. | – | secondhand smoking does not alter STAT3 expression in young hamsters | [72] |
| aged (72-week-old) hamsters with secondhand cigarette smoke exposure (10 cigarettes for 30 min, 4 weeks) | aged hamsters without secondhand cigarette smoke exposure | LV | N.D. | N.D. | N.A. | ↑ (NC) | STAT3 expression showed a tendency of increase in aged hamsters due to secondhand smoking | ||
| Rat Sprague-Dawley Male | young (6-week-old) rats with secondhand cigarette smoke exposure (10 cigarettes for 30 min, twice a day for 4 weeks) | young rats without secondhand cigarette smoke exposure | LV | N.D. | N.D. | N.A. | ↑ | secondhand smoking increases STAT3 expression in young rats | [73] |
| aged (18-month-old) rats with secondhand cigarette smoke exposure (10 cigarettes for 30 min, twice a day for 4 weeks) | aged rats with without secondhand cigarette smoke exposure | LV | N.D. | N.D. | N.A. | ↑ (NC) | STAT3 expression showed a tendency of increase in aged rats due to secondhand smoking | ||
| ALCOHOL | |||||||||
| Mouse Wild-type friendly virus B Male | 4% alcohol liquid diet for 12 weeks | regular liquid diet (without ethanol) | ventricles | ↓ | ↓ | Ser727 | – | chronic 4% alcohol liquid diet decreases STAT3 activation and phosphorylation | [74] |
| Mouse Transgenic overexpressing ALDH2 Male | 4% alcohol liquid diet for 12 weeks | regular liquid diet (without ethanol) | ventricles | – | – | Ser727 | – | chronic 4% alcohol liquid diet does not alter STAT3 activation, phosphorylation and expression in mice overexpressing ALDH2 | |
| wild-type 4% alcohol liquid diet for 12 weeks | ventricles | ↑ | ↑ (NC) | Ser727 | – | chronic 4% alcohol liquid diet increases STAT3 activation and does not alter STAT3 expression | |||
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Mouse C57Bl6/J Female | aged >13 months | young (<3 months) | RV | – (NC) | ↓ | Ser727 | ↓ | age decreases STAT3 phosphorylation and expression | [18] |
| Mouse C57Bl6/J Female/Male | aged 21 months | young (8 weeks) | LV mitochondrial fraction | N.D. | N.D. | N.A. | ↓ | age decreases STAT3 expression | [80] |
| Rat Sprague-Dawley Male | aged 24 months | young (6 months) | LV | ↓ | ↓ (NC) | Tyr705 | – (NC) | age decreases STAT3 activation and reduced STAT3 activation may contribute to age-associated hypertrophy | [81] |
| adult 12 months | ↓ | ↓ (NC) | Tyr705 | – (NC) | |||||
| Mouse C57BL/6J Male | aged 14 months | young (2 months) | whole heart | – | – | Tyr705 | ↑ | age does not alter STAT3 activation and phosphorylation, but increases STAT3 expression | [82] |
| Rat Sprague-Dawley Male | aged 20‒24 months | young (3‒4 months) | whole heart | – | – (NC) | Ser727 | – (NC) | age does not influence STAT3 activation | [83] |
| Mouse C57BL/6J Male | aged 24 months | young (3 months) | whole heart | – (NC) | – | Tyr705 | – | age does not influence STAT3 phosphorylation and expression | [84] |
| Hamster N.A. Male | aged 72 weeks | young (6 weeks) | LV | N.D. | N.D. | N.A. | ↑ | age increases STAT3 expression | [72] |
| Rat Sprague-Dawley Male | aged 18 months | young (6 weeks) | LV | N.D. | N.D. | N.A. | ↑ | age increases STAT3 expression | [73] |
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Rat Sprague-Dawley Male | in vivo regional (LAD) 30 min/120 min I/R in STZ-induced diabetes | I/R nondiabetic | ischaemic zone | ↓ (NC) | ↓ | Tyr705 | – | diabetes decreases post-ischaemic STAT3 phosphorylation | [93] |
| LV | ↓ | ↓ | Tyr705 Ser727 | – (NC) | diabetes decreases post-ischaemic STAT3 activation and phosphorylation | [29] | |||
| whole heart | ↓ | ↓ | Tyr705 | – (NC) | diabetes decreases post-ischaemic STAT3 activation and phosphorylation | [31] | |||
| ventricles | ↓ | ↓ | Tyr705 Ser727 | – | diabetes decreases post-ischaemic STAT3 activation and phosphorylation | [92] | |||
| Rat Sprague-Dawley Male | in vivo regional (LAD) 30 min/90 min I/R in STZ-induced diabetes | I/R nondiabetic | ischaemic zone | ↓ | ↓ | Tyr705 | – (NC) | diabetes decreases post-ischaemic STAT3 activation and phosphorylation | [94] |
| Rat Sprague-Dawley Male | ex vivo global 30 min/120 min I/R in STZ-induced diabetes | I/R nondiabetic | LV | ↓ | ↓ (NC) | Tyr705 | – (NC) | diabetes decreases post-ischaemic STAT3 activation | [33] |
| Mouse leptin receptor null, homozygous db/db Male | ex vivo global 30 min/60 min I/R in high-fat-diet-induced diabetes | I/R C57BL/6J wild-type mouse | whole heart | ↓ | ↓ | Tyr705 | ↓ | diabetes decreases post-ischaemic STAT3 activation, phosphorylation and expression | [95] |
| Rat Sprague-Dawley Male | isolated adult diabetic rat cardiomyocytes subjected to SI/R | isolated adult nondiabetic rat cardiomyocytes subjected to SI/R | cells | ↓ | ↓ | Tyr705 | – (NC) | diabetes decreases post-ischaemic STAT3 activation and phosphorylation | [33] |
| H9c2 cells | high glucose conditions + 6 h/12 h SI/R | normal glucose conditions + 6 h/12 h SI/R | cells | ↓ | ↓ | Tyr705 Ser727 | – (NC) | high glucose condition decreases post-ischaemic STAT3 activation and phosphorylation | [29] |
| high glucose conditions | cells | ↓ | ↓ | Tyr705 | – (NC) | high glucose condition decreases post-ischaemic STAT3 activation and phosphorylation | [32] | ||
| Rat Sprague-Dawley Male | isolated adult rat cardiomyocytes subjected to high glucose conditions + 45 min/2 h SI/R | normal glucose conditions + 45 min/2 h SI/R | cells | ↓ | ↓ (NC) | Tyr705 | – (NC) | high glucose condition decreases post-ischaemic STAT3 activation | [92] |
| H9c2 cells | high glucose conditions + 45 min/2 h SI/R | normal glucose conditions + 45 min/2 h SI/R | cells | ↓ | ↓ (NC) | Tyr705 | – (NC) | ||
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| OBESITY | |||||||||
| Mouse leptin -receptor-deficient (ob/ob) obese Male | leptin-receptor-deficient (ob/ob) obese mice + heart failure induced by coronary artery ligation | non-obese + heart failure induced by coronary artery ligation | whole heart | ↓ (NC) | ↓ | Tyr705 | ↓ | ob/ob obesity decreases STAT3 phosphorylation and expression in heart failure | [58] |
| Rabbit New Zealand white Male | in vivo regional 30 min/10 min I/R in diet-induced hypercholesterolemic rabbits | IR in normo- cholesterolemia | whole heart | – | – (NC) | Tyr705 | – (NC) | diet-induced hypercholesterolemia has no effect on post-ischaemic STAT3 activation | [98] |
| CHRONIC KIDNEY DISEASE | |||||||||
| Rat Wistar Male | in vivo regional (LAD) 25 min/120 min I/R in 5/6 nephrectomy-induced CKD | I/R sham | whole heart | – | – | Tyr705 | – | I/R has no effect on STAT3 in 5/6 nephrectomy-induced CKD | [99] |
| AGING | |||||||||
| Mouse C57Bl6/J Female | in vivo regional 30 min/10 min I/R in aged mice | young I/R | LV | ↓ | ↓ (NC) | Ser727 | – (NC) | age decreases post-ischaemic STAT3 activation | [18] |
| Rat Sprague-Dawley Male | in vivo regional 30 min/15 min I/R in aged rats | young I/R | whole heart | – | – (NC) | Ser727 | – (NC) | age does not influence post-ischaemic STAT3 activation | [83] |
| GENDER | |||||||||
| Mouse C57BL/6 Female/Male | ex vivo global 20 min/60 min I/R in male mice | I/R in female mice | whole heart | ↓ | ↓ | Tyr705 | – (NC) | post-ischaemic STAT3 activation and phosphorylation is lower in male mice | [106] |
| N.D. | N.D. | N.A. | ↓ * | post-ischaemic STAT3 mRNA expression is lower in male mice | |||||
| Rat Sprague-Dawley Female/Male | ex vivo global 25 min/40 min I/R in male mice | I/R in female mice | whole heart | ↓ | ↓ (NC) | Tyr705 | – (NC) | post-ischaemic STAT3 activation is lower in male rats | [107] |
| DEPRESSION | |||||||||
| Rat Sprague-Dawley Male | ex vivo regional (LCA) 35 min/10 min I/R in chronic mild stress (3-week-long)-induced depression | I/R non-depressed | LV | – | – (NC) | Tyr705 | – (NC) | depression does not alter post-ischaemic STAT3 activation | [109] |
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Rat Sprague-Dawley Male | in vivo regional (LAD) 30 min/120 min I/R in STZ-induced diabetes + morphine | I/R nondiabetic + morphine | ischaemic zone | ↓ (NC) | ↓ | Tyr705 | – | diabetes attenuates morphine-induced post-ischaemic STAT3 phosphorylation, and morphine cannot enhance post-ischaemic STAT3 phosphorylation in diabetes, which may contribute to the abrogation of morphine-induced cardioprotection in diabetes | [93] |
| I/R in STZ-induced diabetes | ischaemic zone | – (NC) | – (NC) | Tyr705 | – (NC) | ||||
| ex vivo global 30 min/120 min I/R in STZ-induced diabetes + isoflurane postconditioning (PostC) | I/R nondiabetic + isoflurane PostC | LV | ↓ | ↓ (NC) | Tyr705 | – (NC) | diabetes attenuates post-ischaemic STAT3 activation due to isoflurane PostC, and isoflurane PostC cannot enhance post-ischaemic STAT3 activation, which may contribute to the abrogation of cardioprotection by isoflurane PostC in diabetes | [33] | |
| I/R in STZ-induced diabetes | LV | – | – (NC) | Tyr705 | – (NC) | ||||
| in vivo 30 min/90 min I/R in STZ-induced diabetes + sevoflurane PostC | I/R nondiabetic + sevoflurane PostC | area at risk | ↓ (NC) | ↓ (NC) | Tyr705 | – (NC) | the activation of STAT3 due to sevoflurane PostC is lower when applied in diabetes, and sevoflurane PostC cannot enhance post-ischaemic STAT3 activation both in diabetic rats | [94] | |
| in vivo 30 min/90 min I/R in STZ-induced diabetes | area at risk | – | – (NC) | Tyr705 | – (NC) | ||||
| in vivo 30 min/120 min I/R in STZ-induced diabetes + ischaemic PostC | I/R nondiabetic + ischaemic PostC | whole heart | ↓ (NC) | ↓ (NC) | Tyr705 | – (NC) | diabetes abrogates post-ischaemic STAT3 activation due to ischaemic PostC | [31] | |
| I/R in STZ-induced diabetes | whole heart | – | – (NC) | Tyr705 | – (NC) | ||||
| Rat Sprague-Dawley Male | in vivo 30 min/120 min I/R in STZ-induced diabetes + repeated non-invasive limb ischaemic preconditioning | I/R nondiabetic + repeated non-invasive limb ischaemic PostC | LV | ↓ (NC) | ↓ (NC) | Tyr705 Ser727 | – (NC) | the activation of STAT3 is lower in repeated non-invasive limb ischaemic preconditioning when applied in diabetes, the STAT3 activation is increased due to preconditioning in diabetes | [29] |
| I/R in STZ-induced diabetes | LV | ↑ | ↑ (NC) | Tyr705 Ser727 | – (NC) | ||||
| H9c2 cells | 6 h/12 h SI/R under high glucose conditions + remote time-repeated hypoxic preconditioning | normal glucose condition + remote time-repeated hypoxic preconditioning | cells | ↓ (NC) | ↓ (NC) | Tyr705 Ser727 | – (NC) | the activation of STAT3 is lower in remote time-repeated hypoxic preconditioning when applied in diabetes, the STAT3 activation is increased due to remote time-repeated hypoxic preconditioning in high glucose conditions | [29] |
| high glucose conditions + 6 h/12 h SI/R | cells | ↑ | ↑ | Tyr705 Ser727 | – (NC) | ||||
| Mouse leptin receptor null, homozygous db/db Male | ex vivo global 30 min/60 min I/R in db/db diabetes + rapamycin | I/R nondiabetic + rapamycin | whole heart | – | – | Tyr705 | – | rapamycin increases (restores) STAT3 activation, phosphorylation and expression in diabetes | [95] |
| ex vivo global I/R 30 min/60 min in db/db diabetes | whole heart | ↑ | ↑ | Tyr705 | ↑ | ||||
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Mouse leptin receptor null, homozygous db/db Male | ex vivo global 30 min/60 min I/R in type II diabetes + Rapatar | I/R in type II diabetes | whole heart | ↑ | ↑ | Tyr705 | – (NC) | Rapatar treatment induces post-ischaemic STAT3 phosphorylation in diabetes | [114] |
| Rat Sprague-Dawley Male | in vivo 30 min/90 min I/R in STZ-induced diabetes + N-acetylcysteine | in vivo 30 min/90 min I/R in STZ-induced diabetes | area at risk | ↑ | ↑ (NC) | Tyr705 | – (NC) | N-acetylcysteine enhances post-ischaemic STAT3 activation in diabetes | [94] |
| in vivo 30 min/90 min I/R in STZ-induced diabetes + sevoflurane postconditioning + N-acetylcysteine | in vivo 30 min/90 min I/R in STZ-induced diabetes | area at risk | ↑ | ↑ (NC) | Tyr705 | – (NC) | STAT3 activation induced by sevoflurane postconditioning + N-acetylcysteine is superior to sevoflurane postconditioning or N-acetylcysteine in diabetes | [94] | |
| in vivo 30 min/90 min I/R in STZ-induced diabetes + sevoflurane postconditioning | ↑ | ↑ (NC) | Tyr705 | – (NC) | |||||
| in vivo 30 min/90 min I/R in STZ-induced diabetes + N-acetylcysteine | ↑ | ↑ (NC) | Tyr705 | – (NC) | |||||
| in vivo regional (LAD) 30 min/120 min I/R in STZ-induced diabetes + N-acetylcysteine + allopurinol | I/R in STZ-induced diabetes | ventricles | ↑ | ↑ (NC) | Tyr705 Ser727 | – (NC) | N-acetylcysteine + allopurinol increases (preserves) post-ischaemic STAT3 activation in diabetes | [92] | |
| isolated adult rat cardiomyocytes subjected to 45 min/2 h SI/R under high glucose conditions + adiponectin | high glucose conditions + 45 min/2 h SI/R | cells | ↑ | ↑ (NC) | Tyr705 | – (NC) | adiponectin increases post-ischaemic STAT3 activation in high glucose conditions | ||
| isolated adult rat cardiomyocytes subjected to 45 min/2 h SI/R under high glucose conditions + N-acetylcysteine + allopurinol | ↑ | ↑ (NC) | Tyr705 | – (NC) | N-acetylcysteine + allopurinol increases (preserves) post-ischaemic STAT3 activation in high glucose conditions | ||||
| H9c2 cells | 45 min/2 h SI/R under high glucose conditions + N-acetylcysteine + allopurinol | high glucose conditions + 45 min/2 h SI/R | cells | ↑ | ↑ (NC) | Tyr705 | – (NC) | ||
| 12 h/6 h SI/R under high glucose conditions + propofol | high glucose conditions + 12 h/6 h SI/R | ↑ | ↑ | Tyr705 | – (NC) | propofol enhances STAT3 activation in settings of SI/R under high glucose conditions | [32] | ||
| Animal or Cell | Test Group | Control Group | Tissue Sample | p-STAT3/t-STAT3 Activation | p-STAT3 Phosphorylation | t-STAT3 Expression | Conclusions | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| OBESITY | |||||||||
| Rabbit New Zealand White Male | in vivo regional 30 min/10 min I/R in diet-induced hypercholesterolemic rabbits + oleuropein | I/R in diet-induced hypercholesterolemic rabbits | whole heart | ↑ | ↑ (NC) | Tyr705 | – (NC) | oleuropein increases post-ischaemic STAT3 activation in diet-induced hypercholesterolemia | [98] |
| CHRONIC KIDNEY DISEASE | |||||||||
| Rat Wistar Male | 5/6 nephrectomy-induced CKD + ischaemic preconditioning | I/R in 5/6 nephrectomy-induced CKD | whole heart | ↑ | ↑ | Tyr705 | – | ischaemic preconditioning increases STAT3 activation and phosphorylation in 5/6 nephrectomy-induced CKD | [99] |
| AGING | |||||||||
| Mouse C57Bl6/J Female | in vivo regional I/R + ischaemic postconditioning (PostC) in aged mice | young I/R + ischaemic PostC | LV | ↓ | ↓ (NC) | Ser727 | – (NC) | STAT3 activation is lower in aged rats subjected to ischaemic PostC, which may contribute to the age-related loss of ischaemic PostC-induced protection | [18] |
| Rat Sprague-Dawley Male | in vivo regional I/R + sevoflurane-PostC in aged rats | young I/R + sevoflurane-PostC | whole heart | – | – (NC) | Ser727 | – (NC) | age does not influence STAT3 activation in sevoflurane-PostC | [83] |
| DEPRESSION | |||||||||
| Rat Sprague-Dawley Male | ex vivo regional (LCA) 35 min/10 min I/R in depression induced by chronic mild stress (3-week) + ischaemic PostC | I/R non-depressed + ischaemic PostC | LV | ↓ | ↓ (NC) | Tyr705 | – (NC) | the activation of STAT3 due to ischaemic PostC is abrogated when applied in chronic mild stress | [109] |
| I/R in chronic mild stress-induced depression | LV | – | – (NC) | Tyr705 | – (NC) | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pipicz, M.; Demján, V.; Sárközy, M.; Csont, T. Effects of Cardiovascular Risk Factors on Cardiac STAT3. Int. J. Mol. Sci. 2018, 19, 3572. https://doi.org/10.3390/ijms19113572
Pipicz M, Demján V, Sárközy M, Csont T. Effects of Cardiovascular Risk Factors on Cardiac STAT3. International Journal of Molecular Sciences. 2018; 19(11):3572. https://doi.org/10.3390/ijms19113572
Chicago/Turabian StylePipicz, Márton, Virág Demján, Márta Sárközy, and Tamás Csont. 2018. "Effects of Cardiovascular Risk Factors on Cardiac STAT3" International Journal of Molecular Sciences 19, no. 11: 3572. https://doi.org/10.3390/ijms19113572
APA StylePipicz, M., Demján, V., Sárközy, M., & Csont, T. (2018). Effects of Cardiovascular Risk Factors on Cardiac STAT3. International Journal of Molecular Sciences, 19(11), 3572. https://doi.org/10.3390/ijms19113572