Label-Free Quantitative Proteomics in a Methylmalonyl-CoA Mutase-Silenced Neuroblastoma Cell Line

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. MUT Silencing
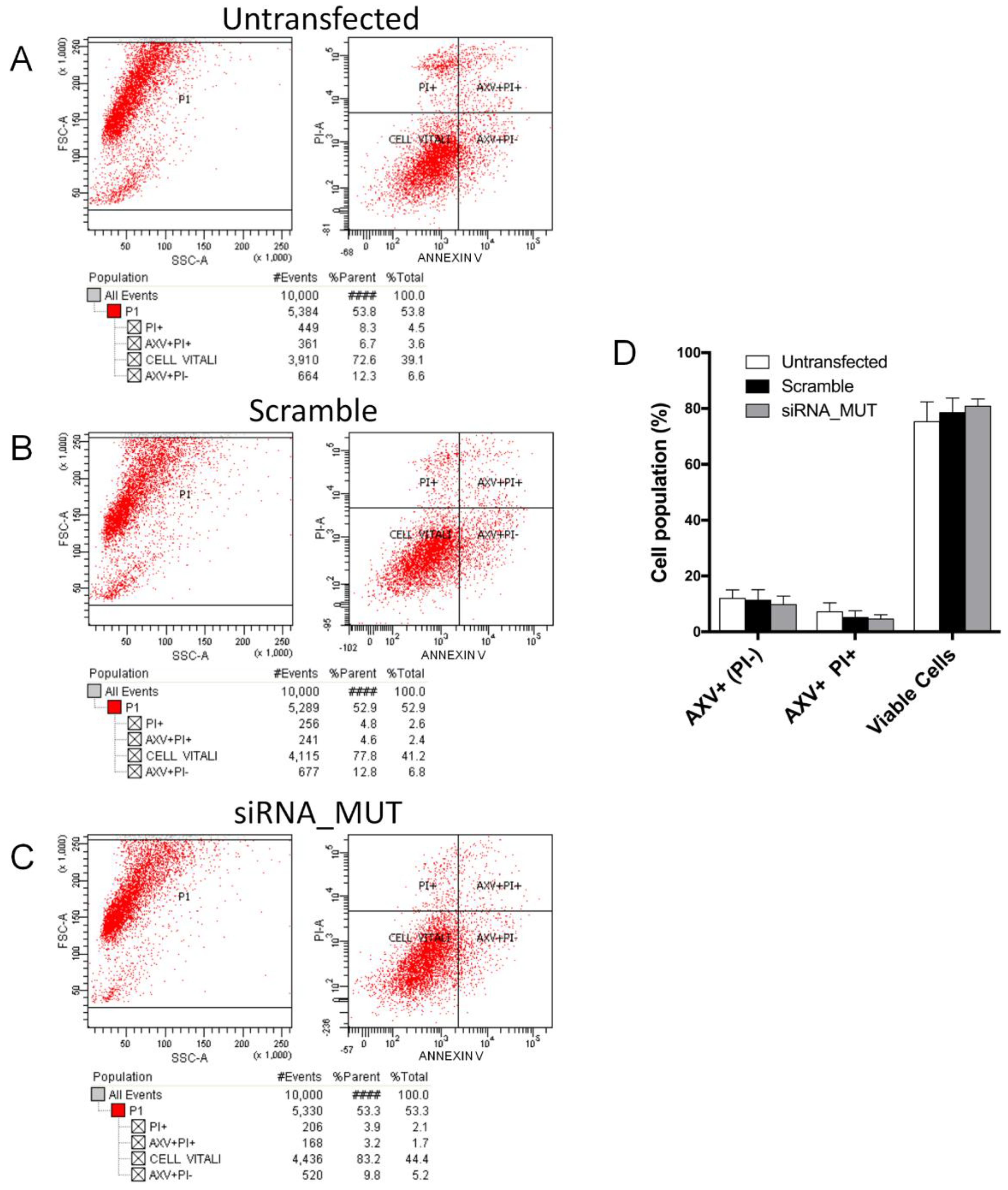
2.2. Cell Survival and Apoptosis
2.3. Proteomic Profiles
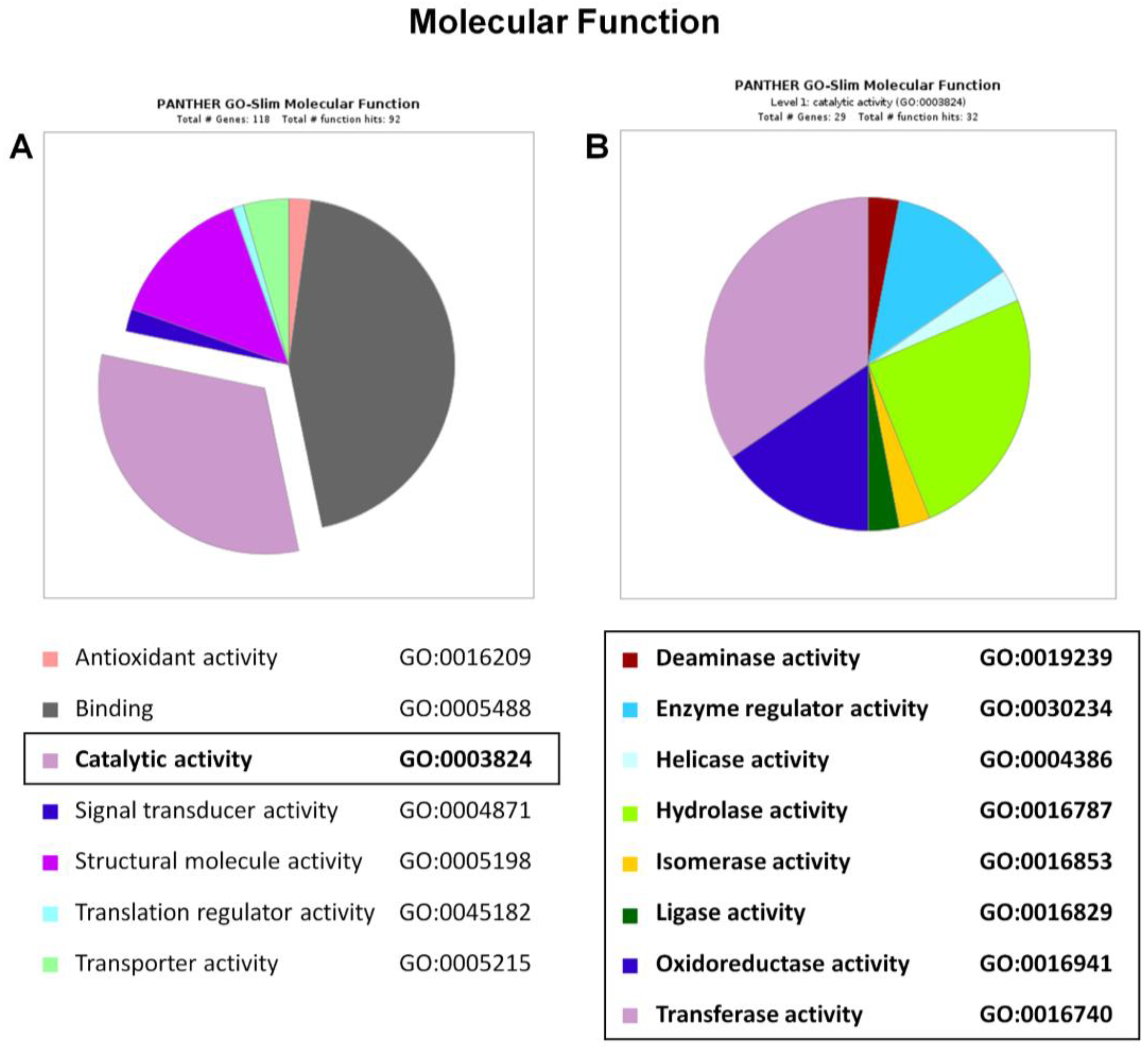
2.4. Functional and Biological Annotation
2.5. MUT Silencing Decreases Cell Viability and Mitochondrial Functionality in Propionate-Enriched Culture Medium
3. Material and Methods
3.1. Cell Culture and Small Interfering RNA Transfection
3.2. Cellular Lysis and Western Blotting Analysis
3.3. Apoptosis Assay by Flow Cytometry
3.4. Neutral-Red and MTT Assays
3.5. Proteomic Analysis
3.6. Quantitative Label-Free Comparative Analysis
3.7. Bioinformatic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fenton, W.A.; Gravel, R.A.; Rosenblatt, D.S. Disorders of propinate and methylmalonate metabolism. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Ruoppolo, M.; Scolamiero, E.; Caterino, M.; Mirisola, V.; Franconi, F.; Campesi, I. Female and male human babies have distinct blood metabolomic patterns. Mol. Biosyst. 2015, 11, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Scolamiero, E.; Villani, G.R.D.; Ingenito, L.; Pecce, R.; Albano, L.; Caterino, M.; di Girolamo, M.G.; Di Stefano, C.; Franzese, I.; Gallo, G.; et al. Maternal vitamin B12 deficiency detected in expanded newborn screening. Clin. Biochem. 2014, 47, 312–317. [Google Scholar] [PubMed]
- Scolamiero, E.; Cozzolino, C.; Albano, L.; Ansalone, A.; Caterino, M.; Corbo, G.; di Girolamo, M.G.; Di Stefano, C.; Durante, A.; Franzese, G.; et al. Targeted metabolomics in the expanded newborn screening for inborn errors of metabolism. Mol. Biosyst. 2015, 11, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Catanzano, F.; Ombrone, D.; Di Stefano, C.; Rossi, A.; Nosari, N.; Scolamiero, E.; Tandurella, I.; Frisso, G.; Parenti, G.; Ruoppolo, M.; et al. The first case of mitochondrial acetoacetyl-CoA thiolase deficiency identified by expanded newborn metabolic screening in Italy: The importance of an integrated diagnostic approach. J. Inherit. Metab. Dis. 2010, 33, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Imperlini, E.; Santorelli, L.; Orru’, S.; Scolamiero, E.; Ruoppolo, M.; Caterino, M. Mass Spectrometry-Based Metabolomic and Proteomic Strategies in Organic Acidemias. Biomed Res. Int. 2016, 2016, 9210408. [Google Scholar] [PubMed]
- Ruoppolo, M.; Caterino, M.; Albano, L.; Pecce, R.; Di Girolamo, M.G.; Crisci, D.; Costanzo, M.; Milella, L.; Franconi, F.; Campesi, I. Targeted metabolomic profiling in rat tissues reveals sex differences. Sci. Rep. 2018, 16, 4663. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Catanzano, F.; Ruoppolo, M.; Alfonsi, L.; Vitale, D.F.; Pecce, R.; Pasanisi, F.; Contaldo, F.; Salvatore, F. Citrulline blood levels as indicators of residual intestinal absorption in patients with short bowel syndrome. Ann. Nutr. Metab. 2008, 53, 137–142. [Google Scholar] [CrossRef] [PubMed]
- de Baulny, H.O.; Benoist, J.F.; Rigal, O.; Touati, G.; Rabier, D.; Saudubray, J.M. Methylmalonic and propionic acidaemias: Management and outcome. J. Inherit. Metab. Dis. 2005, 28, 415–423. [Google Scholar] [PubMed]
- Fraser, J.L.; Venditti, C.P. Methylmalonic and propionic acidemias: Clinical management update. Curr. Opin. Pediatr. 2016, 28, 682–693. [Google Scholar] [PubMed]
- Niemi, A.K.; Kim, I.K.; Krueger, C.E.; Cowan, T.M.; Baugh, N.; Farrell, R.; Bonham, C.A.; Concepcion, W.; Esquivel, C.O.; Enns, G.M. Treatment of methylmalonic acidemia by liver or combined liver-kidney transplantation. J. Pediatr. 2015, 166, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.H.; Hashimoto, T.; Suzuki, T.; Daoud, G.A.; Goto, T.; Nakajima, Y.; Kato, T.; Hibi, M.; Tomishige, H.; Hara, F.; et al. Children undergoing liver transplantation for treatment of inherited metabolic diseases are prone to higher oxidative stress, complement activity and transforming growth factor-b1. Ann. Transplant. 2013, 18, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Vernon, H.J.; Sperati, C.J.; King, J.D.; Poretti, A.; Miller, N.R.; Sloan, J.L.; Cameron, A.M.; Myers, D.; Venditti, C.P.; Valle, D. A detailed analysis of methylmalonic acid kinetics during hemodialysis and after combined liver/kidney transplantation in a patient with mut0 methylmalonic acidemia. J. Inherit. Metab. Dis. 2014, 37, 899–907. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.S.; Theisen, M.; Hong, S.J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2017, 19, 3548–3558. [Google Scholar] [CrossRef] [PubMed]
- Old, W.M.; Meyer-Arendt, K.; Aveline-Wolf, L.; Pierce, K.G.; Mendoza, A.; Sevinsky, J.R.; Resing, K.A.; Ahn, N.G. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell Proteom. 2005, 4, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.; Aspesi, A.; Pavesi, E.; Imperlini, E.; Pagnozzi, D.; Ingenito, L.; Santoro, C.; Dianzani, I.; Ruoppolo, M. Analysis of the interactome of ribosomal protein19 mutants. Proteomics 2014, 20, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Alberio, T.; Pieroni, L.; Ronci, M.; Banfi, C.; Bongarzone, I.; Bottoni, P.; Brioschi, M.; Caterino, M.; Chinello, C.; Cormio, A.; et al. Toward the Standardization of Mitochondrial Proteomics: The Italian Mitochondrial Human Proteome Project Initiative. J. Proteome Res. 2017, 16, 4319–4329. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, V.; Caterino, M.; Iaffaldano, L.; Nardelli, C.; Sirico, A.; Del Vecchio, L.; Martinelli, P.; Pastore, L.; Pucci, P.; Sacchetti, L. Proteome analysis of human amniotic mesenchymal stem cells (hA-MSCs) reveals impaired antioxidant ability, cytoskeleton and metabolic functionality in maternal obesity. Sci. Rep. 2016, 6, 25270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caterino, M.; Zacchia, M.; Costanzo, M.; Bruno, G.; Arcaniolo, D.; Trepiccione, F.; Siciliano, R.A.; Mazzeo, M.F.; Ruoppolo, M.; Capasso, G. Urine Proteomics Revealed a Significant Correlation Between Urine-Fibronectin Abundance and Estimated-GFR Decline in Patients with Bardet-Biedl Syndrome. Kidney Blood Press Res. 2018, 43, 389–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Q.; Xiong, Y.; Fan, L.; Qiao, B.; Xia, Z.; Hu, L.; Wang, Y.; Peng, G.; Ye, Q. Peroxiredoxin 6 attenuates ischemia- and hypoxia-induced liver damage ofbrain-dead donors. Mol. Med. Rep. 2016, 13, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Wajner, M.; Coelho, J.C. Neurological dysfunction in methylmalonic acidaemia is probably related to the inhibitory effect of methylmalonate on brain Energy production. J. Inherit. Metab. Dis. 1997, 20, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Morath, M.A.; Okun, J.G.; Müller, I.B.; Sauer, S.W.; Hörster, F.; Hoffmann, G.F.; Kölker, S. Neurodegeneration and chronic renal failure in methylmalonic aciduria-a pathophysiological approach. J. Inherit. Metab. Dis. 2008, 31, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Melo, D.R.; Kowaltowski, A.J.; Wajner, M.; Castilho, R.F. Mitochondrial Energy metabolism in neurodegeneration associated with methylmalonic acidemia. J. Bioenergy Biomembr. 2011, 43, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; Zerfas, P.M.; Shanske, S.; Sloan, J.; Hoffmann, V.; DiMauro, S.; Venditti, C.P. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J. 2009, 23, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.; Chandler, R.J.; Sloan, J.L.; Dorko, K.; Cusmano-Ozog, K.; Ingenito, L.; Strom, S.C.; Imperlini, E.; Scolamiero, E.; Venditti, C.P.; et al. The proteome of methylmalonic acidemia (MMA): The elucidation of altered pathways in patient livers. Mol. Biosyst. 2016, 12, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Putt, D.A.; Lash, L.H. Enrichment and functional reconstitution of glutathione transport activity from rabbit kidney mitochondria: Further evidence for the role of the dicarboxylate and 2-oxoglutarate carriers in mitochondrial glutathione transport. Arch. Biochem. Biophys. 2000, 373, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.; Pastore, A.; Strozziero, M.G.; Di Giovamberardino, G.; Imperlini, E.; Scolamiero, E.; Ingenito, L.; Boenzi, S.; Ceravolo, F.; Martinelli, D.; et al. The proteome of cblC defect: In vivo elucidation of altered cellular pathways in humans. J. Inherit. Metab. Dis. 2015, 38, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Feldman, G.L.; Poulik, J.; Stockton, D.W.; Sood, B.G. Methylmalonic acidemia presenting as persistent pulmonary hypertension of the newborn. J. Neonatal. Perinat. Med. 2014, 7, 247–251. [Google Scholar]
- Brusque, A.; Rotta, L.; Pettenuzzo, L.F.; Junqueira, D.; Schwarzbold, C.V.; Wyse, A.T.; Wannmacher, C.M.; Dutra-Filho, C.S.; Wajner, M. Chronic postnatal administration of methylmalonic acid provokes a decrease of myelin content and ganglioside N-acetylneuraminic acid concentration in cerebrum of young rats. Braz. J. Med. Biol. Res. 2001, 34, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Garavelli, L.; Santoro, L.; Iori, A.; Gargano, G.; Braibanti, S.; Pedori, S.; Melli, N.; Frattini, D.; Zampini, L.; Galeazzi, T.; et al. Multiple sulfatase deficiency with neonatal manifestation. Ital. J. Pediatr. 2014, 40, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, J.L.; Johnston, J.J.; Manoli, I.; Chandler, R.J.; Krause, C.; Carrillo Carrasco, N.; Chandrasekaran, S.D.; Sysol, J.R.; O’Brien, K.; Hauser, N.S.; et al. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat. Genet. 2011, 43, 883. [Google Scholar] [CrossRef] [PubMed]
- Imperlini, E.; Spaziani, S.; Mancini, A.; Caterino, M.; Buono, P.; Orrù, S. Synergistic effect of DHT and IGF-1 hyperstimulation in human peripheral blood lymphocytes. Proteomics 2015, 15, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Saide, A.; Cagliani, R.; Cantile, M.; Botti, G.; Russo, G. rpL3 promotes the apoptosis of p53 mutated lung cancer cells by down-regulating CBS and NFκB upon 5-FU treatment. Sci. Rep. 2016, 6, 38369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaled, S.Z.; Cevenini, A.; Yazdi, I.K.; Parodi, A.; Evangelopoulos, M.; Corbo, C.; Scaria, S.; Hu, Y.; Haddix, S.G.; Corradetti, B.; et al. One-pot synthesis of pH-responsive hybrid nanogel particles for the intracellular delivery of small interfering RNA. Biomaterials 2016, 87, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.; Ruoppolo, M.; Fulcoli, G.; Huynth, T.; Orrù, S.; Baldini, A.; Salvatore, F. Transcription factor TBX1 overexpression induces downregulation of proteins involved in retinoic acid metabolism: A comparative proteomic analysis. J. Proteome Res. 2009, 8, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Spaziani, S.; Imperlini, E.; Mancini, A.; Caterino, M.; Buono, P.; Orru, S. Insulin like growth factor 1 receptor signaling induced by supraphysiological doses of IGF-1 in human peripheral blood lymphocytes. Proteomics 2014, 14, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Barbarani, G.; Ronchi, A.; Ruoppolo, M.; Santorelli, L.; Steinfelder, R.; Elangovan, S.; Fugazza, C.; Caterino, M. Unravelling pathways downstream Sox6 induction in K562 erythroid cells by proteomic analysis. Sci. Rep. 2017, 7, 14088. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Cevenini, A.; Salvatore, F. Biomarker discovery by proteomics-based approaches for early detection and personalized medicine in colorectal cancer. Proteom. Clin. Appl. 2017, 11, 1600072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Jupe, S.; Fabregat, A.; Hermjakob, H. Expression data analysis with Reactome. Curr. Protoc. Bioinform. 2015, 49, 8–20. [Google Scholar]
- Croft, D. Building models using Reactome pathways as templates. Methods Mol. Biol. 2013, 1021, 273–283. [Google Scholar] [PubMed]
- Caterino, M.; Ruoppolo, M.; Mandola, A.; Costanzo, M.; Orrù, S.; Imperlini, E. Protein protein interaction networks as a new perspective to evaluate distinct functional roles of voltage-dependent anion channel isoforms. Mol. Biosyst. 2017, 13, 2466–2476. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Swiss-Prot Accession | Gene Name | Protein Descriptions | RSC | FoldNSAF |
|---|---|---|---|---|
| Q9Y678 | COPG1 | Coatomer subunit gamma-1 | −4.62 | −6.22 |
| O14776 | TCRG1 | Transcription elongation regulator 1 | −4.62 | −5.89 |
| Q02952 | TCRG1 | A-kinase anchor protein 12 | −4.50 | −5.07 |
| P08865 | RSSA | 40S ribosomal protein SA | −4.37 | −7.53 |
| Q15365 | PCBP1 | Poly(rC)-binding protein 1 | −4.37 | −7.25 |
| Q02978 | M2OM | Mitochondrial 2-oxoglutarate/malate carrier protein | −4.37 | −7.44 |
| P11717 | MPRI | Cation-independent mannose-6-phosphate receptor | −4.37 | −6.89 |
| P62913 | RL11 | 60S ribosomal protein L11 | −4.23 | −4.45 |
| P48643 | TCPE | T-complex protein 1 subunit epsilon | −4.23 | −8.10 |
| O43242 | PSMD3 | 26S proteasome non-ATPase regulatory subunit 3 | −4.23 | −6.50 |
| Q15477 | SKIV2 | Helicase SKI2W | −4.23 | −6.52 |
| P50395 | GDIB | Rab guanosine diphosphate dissociation inhibitor beta | −4.06 | −5.30 |
| Q14240 | IF4A2 | Eukaryotic initiation factor 4A-II | −4.06 | −6.61 |
| Q01433 | AMPD2 | AMP deaminase 2 | −4.06 | −6.74 |
| P35222 | CTNB1 | Catenin beta-1 | −4.06 | −5.63 |
| P04792 | HSPB1 | Heat shock protein beta-1 | −3.88 | −5.80 |
| P00568 | KAD1 | Adenylate kinase isoenzyme 1 | −3.88 | −7.54 |
| Q14232 | EI2BA | Translation initiation factor eIF-2B subunit alpha | −3.88 | −7.62 |
| P09960 | LKHA4 | Leukotriene A-4 hydrolase | −3.88 | −6.96 |
| P43686 | PRS6B | 26S protease regulatory subunit 6B | −3.88 | −5.96 |
| Q14155 | ARHG7 | Rho guanine nucleotide exchange factor 7 | −3.88 | −6.51 |
| Q13045 | FLII | Protein flightless-1 homolog | −3.88 | −5.57 |
| Q14444 | CAPR1 | Caprin-1 | −3.88 | −6.29 |
| P15924 | DESP | Desmoplakin | −3.88 | −6.56 |
| Q96JQ0 | PCD16 | Protocadherin-16 | −3.88 | −5.92 |
| Q9NR31 | SAR1A | guanosine triphosphate (GTP)-binding protein SAR1a | −3.68 | −4.91 |
| Q9Y3B7 | RM11 | 39S ribosomal protein L11, mitochondrial | −3.68 | −5.75 |
| P30041 | PRDX6 | Peroxiredoxin-6 | −3.68 | −3.73 |
| P84103 | SRSF3 | Serine/arginine-rich splicing factor 3 | −3.68 | −3.53 |
| P61020 | RAB5B | Ras-related protein Rab-5B | −3.68 | −7.36 |
| Q13126 | MTAP | S-methyl-5′-thioadenosine phosphorylase | −3.68 | −7.41 |
| P81605 | DCD | Dermcidin | −3.68 | −7.19 |
| O00232 | PSD12 | 26S proteasome non-ATPase regulatory subunit 12 | −3.68 | −7.64 |
| Q9UFN0 | NPS3A | Protein NipSnap homolog 3A | −3.68 | −7.25 |
| P09972 | ALDOC | Fructose-bisphosphate aldolase C | −3.68 | −6.85 |
| Q8NBJ7 | SUMF2 | Sulfatase-modifying factor 2 | −3.68 | −8.21 |
| P09104 | ENOG | Gamma-enolase | −3.68 | −6.16 |
| P13804 | ETFA | Electron transfer flavoprotein subunit alpha, mitochondrial | −3.68 | −7.05 |
| O15173 | PGRC2 | Membrane-associated progesterone receptor component 2 | −3.68 | −6.49 |
| Q8NC51 | PAIRB | Plasminogen activator inhibitor 1 RNA-binding protein | −3.68 | −6.76 |
| Q96HS1 | PGAM5 | Serine/threonine-protein phosphatase PGAM5, mitochondrial | −3.68 | −6.23 |
| Q9UJU6 | DBNL | Drebrin-like protein | −3.68 | −6.61 |
| Q14194 | DPYL1 | Dihydropyrimidinase-related protein 1 | −3.68 | −7.19 |
| Q96PZ0 | PUS7 | Pseudouridylate synthase 7 homolog | −3.68 | −6.32 |
| P62195 | PRS8 | 26S protease regulatory subunit 8 | −3.68 | −6.82 |
| Q14247 | SRC8 | Src substrate cortactin | −3.68 | −6.25 |
| Q9P289 | STK26 | Serine/threonine-protein kinase 26 | −3.68 | −5.83 |
| Q9Y6E0 | STK24 | Serine/threonine-protein kinase 24 | −3.68 | −5.62 |
| O14579 | COPE | Coatomer subunit epsilon | −3.68 | −6.33 |
| Q13330 | MTA1 | Metastasis-associated protein MTA1 | −3.68 | −5.89 |
| Q16401 | PSMD5 | 26S proteasome non-ATPase regulatory subunit 5 | −3.68 | −6.29 |
| Q15075 | EEA1 | Early endosome antigen 1 | −3.68 | −6.20 |
| Q92626 | PXDN | Peroxidasin homolog | −3.68 | −6.73 |
| O60841 | IF2P | Eukaryotic translation initiation factor 5B | −3.68 | −6.02 |
| Q13576 | IQGA2 | Ras GTPase-activating-like protein IQGAP2 | −3.68 | −4.53 |
| Q5VYK3 | ECM29 | Proteasome-associated protein ECM29 homolog | −3.68 | −4.46 |
| Q96GQ7 | DDX27 | Probable ATP-dependent RNA helicase DEAD box (DDX) 27 | 3.73 | 6.71 |
| Q86VM9 | ZCH18 | Zinc finger (ZNF) CCCH domain-containing protein 18 O | 3.73 | 6.46 |
| Q9Y2A7 | NCKP1 | Nck-associated protein 1 | 3.73 | 5.89 |
| Q9BUJ2 | HNRL1 | Heterogeneous nuclear ribonucleoprotein U-like protein 1 | 3.73 | 5.36 |
| Q9Y6K1 | DNM3A | DNA (cytosine-5)-methyltransferase 3A | 3.73 | 5.39 |
| Q9NXE4 | SMPD4 | Sphingomyelin phosphodiesterase 4 | 3.73 | 5.13 |
| Q9BZJ0 | CRNL1 | Crooked neck-like protein 1 | 3.73 | 4.88 |
| Q13523 | PRP4B | Serine/threonine-protein kinase pre-mRNA-processing factor 4 homolog | 3.73 | 5.28 |
| Q8IXT5 | RB12B | RNA-binding protein 12B | 3.73 | 5.19 |
| Q7KZ85 | SPT6H | Transcription elongation factor suppressor of Ty6 | 3.73 | 5.33 |
| O75691 | UTP20 | Small subunit processome component 20 homolog | 3.73 | 5.29 |
| P20340 | RAB6A | Ras-related protein Rab-6A | 3.93 | 5.05 |
| Q13185 | CBX3 | Chromobox protein homolog 3 | 3.93 | 5.05 |
| O14979 | HNRDL | Heterogeneous nuclear ribonucleoprotein D-like | 3.93 | 4.27 |
| Q5BKZ1 | ZN326 | DBIRD complex subunit ZNF326 | 3.93 | 3.58 |
| Q9Y3I0 | RTCB | tRNA-splicing ligase RtcB homolog | 3.93 | 7.54 |
| Q96A65 | EXOC4 | Exocyst complex component 4 | 3.93 | 7.73 |
| Q00325 | MPCP | Phosphate carrier protein, mitochondrial | 3.93 | 6.53 |
| O60282 | KIF5C | Kinesin heavy chain isoform 5C | 3.93 | 6.06 |
| Q68E01 | INT3 | Integrator complex subunit 3 | 3.93 | 6.26 |
| Q13620 | CUL4B | Cullin-4B | 3.93 | 5.32 |
| P51531 | SMCA2 | Probable global transcription activator SNF2L2 | 3.93 | 6.74 |
| O00299 | CLIC1 | Chloride intracellular channel protein 1 | 4.11 | 5.34 |
| P83916 | CBX1 | Chromobox protein homolog 1 | 4.11 | 5.22 |
| Q96E39 | RMXL1 | RNA binding motif protein, X-linked-like-1 | 4.11 | 5.41 |
| Q14978 | NOLC1 | Nucleolar and coiled-body phosphoprotein 1 | 4.11 | 4.61 |
| Q15061 | WDR43 | WD repeat-containing protein 43 | 4.11 | 7.52 |
| Q8WTT2 | NOC3L | Nucleolar complex protein 3 homolog | 4.11 | 7.91 |
| P23921 | RIR1 | Ribonucleoside-diphosphate reductase large subunit | 4.11 | 6.83 |
| O75400 | PR40A | Pre-mRNA-processing factor 40 homolog A | 4.11 | 5.99 |
| P52948 | NUP98 | Nuclear pore complex protein Nup98-Nup96 | 4.11 | 6.03 |
| O75367 | H2AFY | Core histone macro-H2A.1 | 4.27 | 5.79 |
| O43290 | SNUT1 | U4/U6.U5 tri-snRNP-associated protein 1 | 4.27 | 5.81 |
| O00571 | DDX3X | ATP-dependent RNA helicase DDX3X | 4.27 | 5.53 |
| P39023 | RL3 | 60S ribosomal protein L3 | 4.27 | 4.61 |
| Q14690 | RRP5 | Protein RRP5 homolog | 4.27 | 7.07 |
| Q13151 | ROA0 | Heterogeneous nuclear ribonucleoprotein A0 | 4.42 | 5.96 |
| P38919 | IF4A3 | Eukaryotic initiation factor 4A-III | 4.42 | 6.24 |
| Q9UMS6 | SYNP2 | Synaptopodin-2 | 4.42 | 6.95 |
| Q9NYF8 | BCLF1 | Bcl-2-associated transcription factor 1 | 4.42 | 4.74 |
| Q9H0A0 | NAT10 | N-acetyltransferase 10 | 4.42 | 7.51 |
| Q9UKV3 | ACINU | Apoptotic chromatin condensation inducer in the nucleus | 4.42 | 7.08 |
| P68431 | H31 | Histone H3.1 | 4.55 | 5.66 |
| Q8IY81 | SPB1 | pre-rRNA processing protein FTSJ3 | 4.55 | 5.91 |
| Q9H6R4 | NOL6 | Nucleolar protein 6 | 4.55 | 5.76 |
| Q9NVP1 | DDX18 | ATP-dependent RNA helicase DDX18 | 4.55 | 5.37 |
| Q8WUM0 | NU133 | Nuclear pore complex protein Nup133 | 4.67 | 8.81 |
| Q8NI27 | THOC2 | THO complex subunit 2 | 4.67 | 6.17 |
| P28331 | NDUS1 | Nicotinamide adenine dinucleotide-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial | 4.78 | 5.73 |
| P07197 | NFM | Neurofilament medium polypeptide | 4.78 | 6.51 |
| Q9UIG0 | BAZ1B | Tyrosine-protein kinase bromodomain adjacent to ZNF 1B | 4.78 | 5.85 |
| P62805 | H4 | Histone H4 | 4.88 | 5.38 |
| O00159 | MYO1C | Unconventional myosin-Ic | 4.98 | 6.63 |
| Q9H583 | HEAT1 | HEAT repeat-containing protein 1 | 4.98 | 6.30 |
| P28370 | SMCA1 | Probable global transcription activator SNF2L1 | 5.24 | 5.60 |
| P49792 | RBP2 | E3 small ubiquitin-like modifier-protein ligase RanBP2 | 5.31 | 9.56 |
| P0C0S5 | H2AZ | Histone H2A.Z | 5.38 | 6.29 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costanzo, M.; Cevenini, A.; Marchese, E.; Imperlini, E.; Raia, M.; Del Vecchio, L.; Caterino, M.; Ruoppolo, M. Label-Free Quantitative Proteomics in a Methylmalonyl-CoA Mutase-Silenced Neuroblastoma Cell Line. Int. J. Mol. Sci. 2018, 19, 3580. https://doi.org/10.3390/ijms19113580
Costanzo M, Cevenini A, Marchese E, Imperlini E, Raia M, Del Vecchio L, Caterino M, Ruoppolo M. Label-Free Quantitative Proteomics in a Methylmalonyl-CoA Mutase-Silenced Neuroblastoma Cell Line. International Journal of Molecular Sciences. 2018; 19(11):3580. https://doi.org/10.3390/ijms19113580
Chicago/Turabian StyleCostanzo, Michele, Armando Cevenini, Emanuela Marchese, Esther Imperlini, Maddalena Raia, Luigi Del Vecchio, Marianna Caterino, and Margherita Ruoppolo. 2018. "Label-Free Quantitative Proteomics in a Methylmalonyl-CoA Mutase-Silenced Neuroblastoma Cell Line" International Journal of Molecular Sciences 19, no. 11: 3580. https://doi.org/10.3390/ijms19113580
APA StyleCostanzo, M., Cevenini, A., Marchese, E., Imperlini, E., Raia, M., Del Vecchio, L., Caterino, M., & Ruoppolo, M. (2018). Label-Free Quantitative Proteomics in a Methylmalonyl-CoA Mutase-Silenced Neuroblastoma Cell Line. International Journal of Molecular Sciences, 19(11), 3580. https://doi.org/10.3390/ijms19113580