Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors

 , , , ,
, , , ,
Abstract
: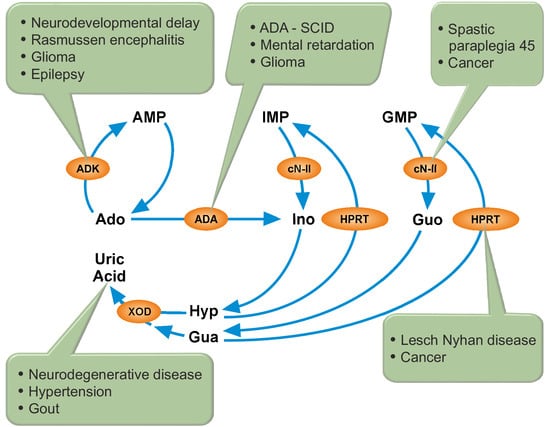
{kind=link}
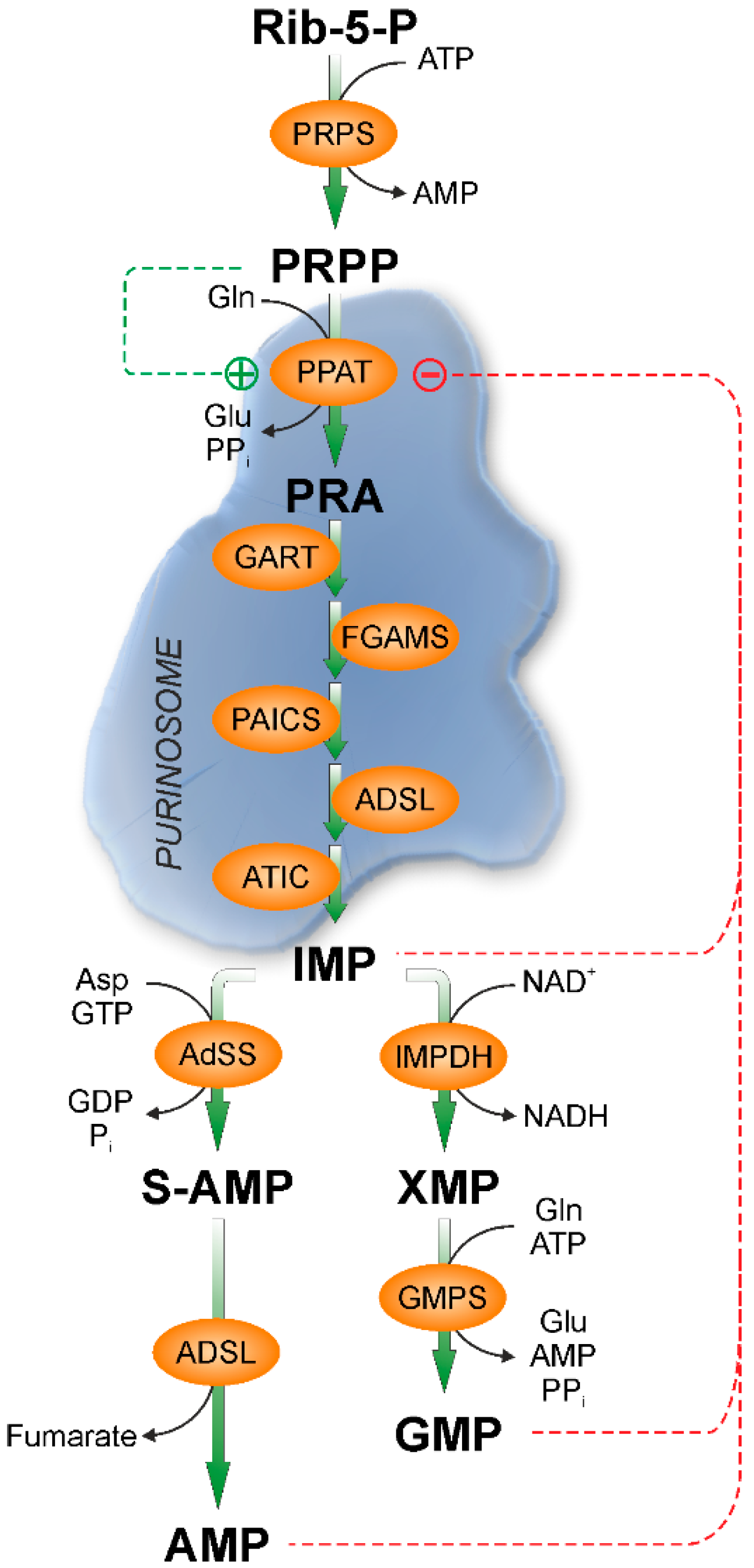{kind=link}
{kind=link}
1. Introduction
2. Cytosolic 5′-Nucleotidase II (cN-II)
2.1. CN-II and Spastic Paraplegia
2.2. CN-II and Cancer
2.3. CN-II and Other Pathologies
3. Adenosine Kinase (ADK) and Adenosine Deaminase (ADA)
3.1. ADK
3.2. ADK and Neurological Diseases
3.3. ADK and ADA in Glioma
3.4. ADA
3.5. ADA and Adenosine Receptors
3.6. ADA Inhibition and Apoptosis
4. Hypoxanthine-Guanine Phosphoribosyltransferase (HPRT)
4.1. HPRT and LND
4.2. HPRT Deficiency and the Accumulation of Potentially Toxic Metabolites
4.3. HPRT and the Formation of Purinosome
4.4. HPRT and the Purinergic Signaling
4.5. HPRT and Cancer
5. Xanthine Oxidase (XOD)
5.1. XOD and Xanthine Dehydrogenase (XDH)
5.2. XOD and Uric Acid
5.3. Pathologies Associated with High Uricemia
5.4. Pathologies Associated with Low Uricemia
6. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| A1R | adenosine receptor subtype 1 |
| A2R | adenosine receptor subtype 2 |
| A3R | adenosine receptor subtype 3 |
| AD | Alzheimer’s disease |
| ADA | adenosine deaminase |
| ADK | adenosine kinase |
| AICAR | 5-aminoimidazole-4-carboxamide ribonucleotide |
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| AMP | adenosine 5′-monophosphate |
| AMPK | AMP-activated protein kinase |
| ALS | amyotrophic lateral sclerosis |
| ATIC | AICAR formyltransferase/IMP cyclohydrolase |
| Ascl1 | achaete-scute family bHLH transcription factor 1 |
| BDNF | TrkB brain-derived neurotrophic factor |
| CD26 | trimeric complexes dipeptidyl peptidase IV |
| CLL | chronic lymphoid leukemia |
| CN-I | cytosolic 5′-nucleotidase-I |
| CN-II | cytosolic 5′-nucleotidase II |
| CNS | central nervous system |
| Darpp-32 | the gene encoding dopamine- and cAMP-regulated phosphoprotein 32 |
| dCF | deoxycoformycin |
| ENT2 | adenosine equilibrative transporters NBTI-insensitive |
| ES | embryonic stem cells |
| Foxp1 | forkhead box P1 |
| GMP | guanosine 5′-monophosphate |
| HPRT | the gene coding for HPRT |
| HPRT | hypoxanthine guanine phosphoribosyl transferase |
| HSPs | hereditary spastic paraplegias |
| IMP | inosine 5′-monophosphate |
| IMPDH | inosine-5′-monophosphate dehydrogenase |
| Ipaf | ice protease-activating factor |
| LND | Lesch–Nyhan disease |
| MAPK | mitogen activated protein kinases |
| 6-MP | 6-mercaptopurine |
| NT5C2 | the gene coding for cN-II |
| NTPDases | 5′-triphosphate diphosphohydrolases |
| NTPase | nucleoside 5-triphosphatase |
| PPAT | 5-phosphoribosyl-1-pyrophosphate amidotransferase |
| PRPP | 5-phosphoribosyl-1-pyrophosphate |
| P2XR | purinergic receptor subtype 2X |
| P2YR | purinergic receptor subtype 2Y |
| SAH | S-adenosylhomocysteine |
| SAHH | SAH hydrolase |
| SCID | severe combined immunodeficiency |
| SNPs | single nucleotide polymorphism |
| SP45 | autosomic recessive spastic paraplegia 45 |
| WNT4 | wingless-type MMTV integration site family, member 4 |
| XDH | xanthine dehydrogenase |
| XOD | xanthine oxidase |
References
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipata, P.L.; Balestri, F.; Camici, M.; Tozzi, M.G. Molecular mechanisms of nucleoside recycling in the brain. Int. J. Biochem. Cell Biol. 2011, 43, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A.; Saha, A.; Chan, A.; Casteel, D.E.; Pilz, R.B.; Boss, G.R. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem. J. 2013, 454, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 2008, 320, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Smith, J.L.; Zalkin, H. Binding of purine nucleotides to two regulatory sites results in synergistic feedback inhibition of glutamine 5-phosphoribosylpyrophosphate amidotransferase. J. Biol. Chem. 1994, 269, 6784–6789. [Google Scholar] [PubMed]
- Boer, P.; Sperling, O. Role of cellular ribose-5-phosphate content in the regulation of 5-phosphoribosyl-1-pyrophosphate and de novo purine synthesis in a human hepatoma cell line. Metabolism 1995, 44, 1469–1474. [Google Scholar] [CrossRef]
- Camici, M.; Allegrini, S.; Tozzi, M.G. Interplay between adenylate metabolizing enzymes and AMP-activated protein kinase. FEBS J. 2018, 285, 3337–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozzi, M.G.; Pesi, R.; Allegrini, S. On the physiological role of cytosolic 5′-nucleotidase II (cN-II): Pathological and therapeutical implications. Curr. Med. Chem. 2013, 20, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Ipata, P.L.; Tozzi, M.G. Recent advances in structure and function of cytosolic IMP-GMP specific 5′-nucleotidase II (cN-II). Purinergic Signal 2006, 2, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Barsotti, C.; Lutzemberger, L.; Camici, M.; Ipata, P.L. Key role of uridine kinase and uridine phosphorylase in the homeostatic regulation of purine and pyrimidine salvage in brain. Neurochem. Int. 2007, 51, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Pesi, R.; Petrotto, E.; Colombaioni, L.; Allegrini, S.; Garcia-Gil, M.; Camici, M.; Jordheim, L.P.; Tozzi, M.G. Cytosolic 5′-Nucleotidase II Silencing in a Human Lung Carcinoma Cell Line Opposes Cancer Phenotype with a Concomitant Increase in p53 Phosphorylation. Int. J. Mol. Sci. 2018, 19, 2115. [Google Scholar] [CrossRef] [PubMed]
- Bricard, G.; Cadassou, O.; Cassagnes, L.E.; Cros-Perrial, E.; Payen-Gay, L.; Puy, J.Y.; Lefebvre-Tournier, I.; Tozzi, M.G.; Dumontet, C.; Jordheim, L.P. The cytosolic 5′-nucleotidase cN-II lowers the adaptability to glucose deprivation in human breast cancer cells. Oncotarget 2017, 8, 67380–67393. [Google Scholar] [CrossRef] [PubMed]
- Cividini, F.; Cros-Perrial, E.; Pesi, R.; Machon, C.; Allegrini, S.; Camici, M.; Dumontet, C.; Jordheim, L.P.; Tozzi, M.G. Cell proliferation and drug sensitivity of human glioblastoma cells are altered by the stable modulation of cytosolic 5′-nucleotidase II. Int. J. Biochem. Cell Biol. 2015, 65, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Cividini, F.; Filoni, D.N.; Pesi, R.; Allegrini, S.; Camici, M.; Tozzi, M.G. IMP-GMP specific cytosolic 5′-nucleotidase regulates nucleotide pool and prodrug metabolism. Biochim. Biophys. Acta 2015, 1850, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, C.; Tozzi, M.G.; Dumontet, C.; Jordheim, L.P. The druggability of intracellular nucleotide-degrading enzymes. Cancer Chemother. Pharmacol. 2016, 77, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Ipata, P.L.; Camici, M.; Micheli, V.; Tozz, M.G. Metabolic network of nucleosides in the brain. Curr. Top. Med. Chem. 2011, 11, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Karlsson, A. Cloning and expression of human deoxyguanosine kinase cDNA. Proc. Natl. Acad. Sci. USA 1996, 93, 7258–7262. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, C.; Tozzi, M.G.; Ipata, P.L. Purine and pyrimidine salvage in whole rat brain. Utilization of ATP-derived ribose-1-phosphate and 5-phosphoribosyl-1-pyrophosphate generated in experiments with dialyzed cell-free extracts. J. Biol. Chem. 2002, 277, 9865–9869. [Google Scholar] [CrossRef] [PubMed]
- Schulte, G.; Fredholm, B.B. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal 2003, 15, 813–827. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. Adenosine as a neuromodulator in neurological diseases. Curr. Opin. Pharmacol. 2008, 8, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illes, P.; Verkhratsky, A. Purinergic neurone-glia signalling in cognitive-related pathologies. Neuropharmacology 2016, 104, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, M.; Lecca, D.; Abbracchio, M.P.; Ceruti, S. Pathophysiological Role of Purines and Pyrimidines in Neurodevelopment: Unveiling New Pharmacological Approaches to Congenital Brain Diseases. Front. Pharmacol. 2017, 8, 941. [Google Scholar] [CrossRef] [PubMed]
- Yue, N.; Huang, H.; Zhu, X.; Han, Q.; Wang, Y.; Li, B.; Liu, Q.; Wu, G.; Zhang, Y.; Yu, J. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J Neuroinflamm. 2017, 14, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A. Recent Advances in CNS P2X7 Physiology and Pharmacology: Focus on Neuropsychiatric Disorders. Front. Pharmacol. 2018, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Giacomelli, A.; Naaldijk, Y.; Sarda-Arroyo, L.; Goncalves, M.C.B.; Correa-Velloso, J.; Pillat, M.M.; de Souza, H.D.N.; Ulrich, H. Purinergic Receptors in Neurological Diseases With Motor Symptoms: Targets for Therapy. Front. Pharmacol. 2018, 9, 325. [Google Scholar] [CrossRef] [PubMed]
- Camici, M.; Garcia-Gil, M.; Tozzi, M.G. The Inside Story of Adenosine. Int. J. Mol. Sci. 2018, 19, 784. [Google Scholar] [CrossRef] [PubMed]
- Waring, W.S. Uric acid: An important antioxidant in acute ischaemic stroke. QJM 2002, 95, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Kutzing, M.K.; Firestein, B.L. Altered uric acid levels and disease states. J. Pharmacol. Exp. Ther. 2008, 324, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pasalic, D.; Marinkovic, N.; Feher-Turkovic, L. Uric acid as one of the important factors in multifactorial disorders--facts and controversies. Biochem. Med. 2012, 22, 63–75. [Google Scholar] [CrossRef]
- Cividini, F.; Tozzi, M.G.; Galli, A.; Pesi, R.; Camici, M.; Dumontet, C.; Jordheim, L.P.; Allegrini, S. Cytosolic 5′-nucleotidase II interacts with the leucin rich repeat of NLR family member Ipaf. PLoS ONE 2015, 10, e0121525. [Google Scholar] [CrossRef] [PubMed]
- Dursun, U.; Koroglu, C.; Kocasoy Orhan, E.; Ugur, S.A.; Tolun, A. Autosomal recessive spastic paraplegia (SPG45) with mental retardation maps to 10q24.3-q25.1. Neurogenetics 2009, 10, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, M.F.; Ibrahim, K.; Chalhoub, N.; Elsotouhy, A.; El Mudehki, N.; Abdel Aleem, A. NT5C2 novel splicing variant expands the phenotypic spectrum of Spastic Paraplegia (SPG45): Case report of a new member of thin corpus callosum SPG-Subgroup. BMC Med. Genet. 2017, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Straussberg, R.; Onoufriadis, A.; Konen, O.; Zouabi, Y.; Cohen, L.; Lee, J.Y.W.; Hsu, C.K.; Simpson, M.A.; McGrath, J.A. Novel homozygous missense mutation in NT5C2 underlying hereditary spastic paraplegia SPG45. Am. J. Med. Genet. A 2017, 173, 3109–3113. [Google Scholar] [CrossRef] [PubMed]
- Darvish, H.; Azcona, L.J.; Tafakhori, A.; Ahmadi, M.; Ahmadifard, A.; Paisan-Ruiz, C. Whole genome sequencing identifies a novel homozygous exon deletion in the NT5C2 gene in a family with intellectual disability and spastic paraplegia. NPJ Genom. Med. 2017, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesi, R.; Micheli, V.; Jacomelli, G.; Peruzzi, L.; Camici, M.; Garcia-Gil, M.; Allegrini, S.; Tozzi, M.G. Cytosolic 5′-nucleotidase hyperactivity in erythrocytes of Lesch-Nyhan syndrome patients. Neuroreport 2000, 11, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Chaloin, L. Therapeutic perspectives for cN-II in cancer. Curr. Med. Chem. 2013, 20, 4292–4303. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Kirstein, M.N.; Khatri, A.; Skubitz, K.M.; Dudek, A.Z.; Greeno, E.W.; Kratzke, R.A.; Lamba, J.K. Pathway-based pharmacogenomics of gemcitabine pharmacokinetics in patients with solid tumors. Pharmacogenomics 2012, 13, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Puy, J.Y.; Cros-Perrial, E.; Peyrottes, S.; Lefebvre, I.; Perigaud, C.; Dumontet, C. Determination of the enzymatic activity of cytosolic 5′-nucleotidase cN-II in cancer cells: Development of a simple analytical method and related cell line models. Anal. Bioanal. Chem. 2015, 407, 5747–5758. [Google Scholar] [CrossRef] [PubMed]
- Tzoneva, G.; Dieck, C.L.; Oshima, K.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Madubata, C.J.; Khiabanian, H.; Yu, J.; Waanders, E.; Iacobucci, I.; et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 2018, 553, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Hnizda, A.; Fabry, M.; Moriyama, T.; Pachl, P.; Kugler, M.; Brinsa, V.; Ascher, D.B.; Carroll, W.L.; Novak, P.; Zaliova, M.; et al. Relapsed acute lymphoblastic leukemia-specific mutations in NT5C2 cluster into hotspots driving intersubunit stimulation. Leukemia 2018, 32, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Dieck, C.L.; Tzoneva, G.; Forouhar, F.; Carpenter, Z.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Kirschner-Schwabe, R.; Lew, S.; Seetharaman, J.; Tong, L.; et al. Structure and Mechanisms of NT5C2 Mutations Driving Thiopurine Resistance in Relapsed Lymphoblastic Leukemia. Cancer Cell 2018, 34, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Wallden, K.; Nordlund, P. Structural basis for the allosteric regulation and substrate recognition of human cytosolic 5′-nucleotidase II. J. Mol. Biol. 2011, 408, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Studer, F.E.; Fedele, D.E.; Marowsky, A.; Schwerdel, C.; Wernli, K.; Vogt, K.; Fritschy, J.M.; Boison, D. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 2006, 142, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.A.; Singh, B.; Park, J.; Gupta, R.S. Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus. Biochem. Biophys. Res. Commun. 2009, 388, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.Y.; Lusardi, T.A.; Williams-Karnesky, R.L.; Lan, J.Q.; Poulsen, D.J.; Boison, D. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J. Cereb. Blood Flow Metab. 2011, 31, 1648–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiese, K.; Jablonski, J.; Boison, D.; Kobow, K. Dynamic Regulation of the Adenosine Kinase Gene during Early Postnatal Brain Development and Maturation. Front. Mol. Neurosci. 2016, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- McNally, T.; Helfrich, R.J.; Cowart, M.; Dorwin, S.A.; Meuth, J.L.; Idler, K.B.; Klute, K.A.; Simmer, R.L.; Kowaluk, E.A.; Halbert, D.N. Cloning and expression of the adenosine kinase gene from rat and human tissues. Biochem. Biophys. Res. Commun. 1997, 231, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Motazacker, M.M.; Garshasbi, M.; Kahrizi, K.; Tzschach, A.; Chen, W.; Behjati, F.; Hadavi, V.; Nieh, S.E.; Abedini, S.S.; et al. Homozygosity mapping in consanguineous families reveals extreme heterogeneity of non-syndromic autosomal recessive mental retardation and identifies 8 novel gene loci. Hum. Genet. 2007, 121, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Bjursell, M.K.; Blom, H.J.; Cayuela, J.A.; Engvall, M.L.; Lesko, N.; Balasubramaniam, S.; Brandberg, G.; Halldin, M.; Falkenberg, M.; Jakobs, C.; et al. Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am. J. Hum. Genet. 2011, 89, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Staufner, C.; Lindner, M.; Dionisi-Vici, C.; Freisinger, P.; Dobbelaere, D.; Douillard, C.; Makhseed, N.; Straub, B.K.; Kahrizi, K.; Ballhausen, D.; et al. Adenosine kinase deficiency: Expanding the clinical spectrum and evaluating therapeutic options. J. Inherit. Metab. Dis. 2016, 39, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Shakiba, M.; Mahjoub, F.; Fazilaty, H.; Rezagholizadeh, F.; Shakiba, A.; Ziadlou, M.; Gahl, W.A.; Behnam, B. Adenosine kinase deficiency with neurodevelopemental delay and recurrent hepatic dysfunction: A case report. Adv. Rare Dis. 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Boison, D. Adenosinergic signaling in epilepsy. Neuropharmacology 2016, 104, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, M.; Iyer, A.; Zurolo, E.; Anink, J.; Heimans, J.J.; Boison, D.; Reijneveld, J.C.; Aronica, E. Overexpression of ADK in human astrocytic tumors and peritumoral tissue is related to tumor-associated epilepsy. Epilepsia 2012, 53, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; He, Y.; Chen, M.; Du, J.; Li, G.; Li, S.; Liu, W.; Long, X. Adenosine deaminase and adenosine kinase expression in human glioma and their correlation with gliomaassociated epilepsy. Mol. Med. Rep. 2015, 12, 6509–6516. [Google Scholar] [CrossRef] [PubMed]
- Luan, G.; Gao, Q.; Guan, Y.; Zhai, F.; Zhou, J.; Liu, C.; Chen, Y.; Yao, K.; Qi, X.; Li, T. Upregulation of adenosine kinase in Rasmussen encephalitis. J. Neuropathol. Exp. Neurol. 2013, 72, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Luan, G.; Wang, X.; Gao, Q.; Guan, Y.; Wang, J.; Deng, J.; Zhai, F.; Chen, Y.; Li, T. Upregulation of Neuronal Adenosine A1 Receptor in Human Rasmussen Encephalitis. J. Neuropathol. Exp. Neurol. 2017, 76, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Luan, G.; Gao, Q.; Zhai, F.; Zhou, J.; Liu, C.; Chen, Y.; Li, T. Adenosine kinase expression in cortical dysplasia with balloon cells: Analysis of developmental lineage of cell types. J. Neuropathol. Exp. Neurol. 2015, 74, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Sisodiya, S.M.; Fauser, S.; Cross, J.H.; Thom, M. Focal cortical dysplasia type II: Biological features and clinical perspectives. Lancet Neurol. 2009, 8, 830–843. [Google Scholar] [CrossRef]
- Boison, D.; Aronica, E. Comorbidities in Neurology: Is adenosine the common link? Neuropharmacology 2015, 97, 18–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Ren, G.; Lusardi, T.; Wilz, A.; Lan, J.Q.; Iwasato, T.; Itohara, S.; Simon, R.P.; Boison, D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J. Clin. Investig. 2008, 118, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, P.; McGarrity, S.; Shen, H.Y.; Boison, D.; Yee, B.K. Working memory and the homeostatic control of brain adenosine by adenosine kinase. Neuroscience 2012, 213, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandau, U.S.; Colino-Oliveira, M.; Jones, A.; Saleumvong, B.; Coffman, S.Q.; Liu, L.; Miranda-Lourenco, C.; Palminha, C.; Batalha, V.L.; Xu, Y.; et al. Adenosine Kinase Deficiency in the Brain Results in Maladaptive Synaptic Plasticity. J. Neurosci. 2016, 36, 12117–12128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams-Karnesky, R.L.; Sandau, U.S.; Lusardi, T.A.; Lytle, N.K.; Farrell, J.M.; Pritchard, E.M.; Kaplan, D.L.; Boison, D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Investig. 2013, 123, 3552–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S-adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. J. Nutr. 2002, 132, 2361S–2366S. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.L.; Ritter, A.C.; Jackson, E.K.; Conley, Y.P.; Kochanek, P.M.; Boison, D.; Wagner, A.K. Genetic variation in the adenosine regulatory cycle is associated with posttraumatic epilepsy development. Epilepsia 2015, 56, 1198–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poppe, D.; Doerr, J.; Schneider, M.; Wilkens, R.; Steinbeck, J.A.; Ladewig, J.; Tam, A.; Paschon, D.E.; Gregory, P.D.; Reik, A.; et al. Genome Editing in Neuroepithelial Stem Cells to Generate Human Neurons with High Adenosine-Releasing Capacity. Stem Cells Transl. Med. 2018, 7, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sai, K.; Yang, D.; Yamamoto, H.; Fujikawa, H.; Yamamoto, S.; Nagata, T.; Saito, M.; Yamamura, T.; Nishizaki, T. A(1) adenosine receptor signal and AMPK involving caspase-9/-3 activation are responsible for adenosine-induced RCR-1 astrocytoma cell death. Neurotoxicology 2006, 27, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, S.; Nagata, K.; Nakahata, N. Adenosine uptake-dependent C6 cell growth inhibition. Eur. J. Pharmacol. 2007, 577, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Torres, A.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martin, R.; Quezada, C. The Adenosine A(3) Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Vargas, Y.; Uribe, D.; Jaramillo, C.; Gleisner, A.; Salazar-Onfray, F.; Lopez, M.N.; Melo, R.; Oyarzun, C.; San Martin, R.; et al. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget 2016, 7, 67373–67386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebel, A.M.; Piato, A.L.; Schaefer, I.C.; Nery, L.R.; Bogo, M.R.; Bonan, C.D. Antiepileptic drugs prevent changes in adenosine deamination during acute seizure episodes in adult zebrafish. Pharmacol. Biochem. Behav. 2013, 104, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Wahlman, C.; Doyle, T.M.; Little, J.W.; Luongo, L.; Janes, K.; Chen, Z.; Esposito, E.; Tosh, D.K.; Cuzzocrea, S.; Jacobson, K.A.; et al. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain 2018, 159, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, K.V.; Gaspar, H.B. Adenosine Deaminase Deficiency—More Than Just an Immunodeficiency. Front. Immunol. 2016, 7, 314. [Google Scholar] [CrossRef] [PubMed]
- Flinn, A.M.; Gennery, A.R. Adenosine deaminase deficiency: A review. Orphanet J. Rare Dis. 2018, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.H.; Lwin, R.; Fairbanks, L.; Gerritsen, B.; Gaspar, H.B. Cognitive and behavioral abnormalities in adenosine deaminase deficient severe combined immunodeficiency. J. Pediatr. 2001, 139, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Nofech-Mozes, Y.; Blaser, S.I.; Kobayashi, J.; Grunebaum, E.; Roifman, C.M. Neurologic abnormalities in patients with adenosine deaminase deficiency. Pediatr Neurol 2007, 37, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Titman, P.; Pink, E.; Skucek, E.; O’Hanlon, K.; Cole, T.J.; Gaspar, J.; Xu-Bayford, J.; Jones, A.; Thrasher, A.J.; Davies, E.G.; et al. Cognitive and behavioral abnormalities in children after hematopoietic stem cell transplantation for severe congenital immunodeficiencies. Blood 2008, 112, 3907–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbs, G.; Litt, M.; Lis, E.; Jackson, R.; Voth, W.; Lindberg, A.; Litt, R. Adenosine deaminase activity decreased in autism. J. Am. Acad. Child Psychiatry 1982, 21, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; De Luca, D.; Saccucci, P.; Fiumara, A.; Elia, M.; Porfirio, M.C.; Lucarelli, P.; Curatolo, P. Autism: Evidence of association with adenosine deaminase genetic polymorphism. Neurogenetics 2001, 3, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Saccucci, P.; Arpino, C.; Rizzo, R.; Gagliano, A.; Volzone, A.; Lalli, C.; Galasso, C.; Curatolo, P. Association of adenosine deaminase polymorphism with mild mental retardation. J. Child Neurol. 2006, 21, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Honig, M.; Albert, M.H.; Schulz, A.; Sparber-Sauer, M.; Schutz, C.; Belohradsky, B.; Gungor, T.; Rojewski, M.T.; Bode, H.; Pannicke, U.; et al. Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of CNS complications. Blood 2007, 109, 3595–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, C.; Gaspar, H.B. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biologics 2009, 3, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, M.P.; Ferrua, F.; Castagnaro, L.; Pajno, R.; Barzaghi, F.; Giannelli, S.; Dionisio, F.; Brigida, I.; Bonopane, M.; Casiraghi, M.; et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood 2016, 128, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, A.V.; Hernandez, R.J.; Fumagalli, F.; Bianchi, V.; Poliani, P.L.; Dallatomasina, C.; Riboni, E.; Politi, L.S.; Tabucchi, A.; Carlucci, F.; et al. Alterations in the brain adenosine metabolism cause behavioral and neurological impairment in ADA-deficient mice and patients. Sci. Rep. 2017, 7, 40136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, P.; Brenhouse, H.C. Broken or maladaptive? Altered trajectories in neuroinflammation and behavior after early life adversity. Dev. Cognit. Neurosci. 2015, 11, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M.R.; Datta, S.K.; Kellems, R.E. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J. Biol. Chem. 1998, 273, 5093–5100. [Google Scholar] [CrossRef] [PubMed]
- Ledent, C.; Vaugeois, J.M.; Schiffmann, S.N.; Pedrazzini, T.; El Yacoubi, M.; Vanderhaeghen, J.J.; Costentin, J.; Heath, J.K.; Vassart, G.; Parmentier, M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature 1997, 388, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ciruela, F.; Albergaria, C.; Soriano, A.; Cuffi, L.; Carbonell, L.; Sanchez, S.; Gandia, J.; Fernandez-Duenas, V. Adenosine receptors interacting proteins (ARIPs): Behind the biology of adenosine signaling. Biochim. Biophys. Acta 2010, 1798, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gracia, E.; Perez-Capote, K.; Moreno, E.; Barkesova, J.; Mallol, J.; Lluis, C.; Franco, R.; Cortes, A.; Casado, V.; Canela, E.I. A2A adenosine receptor ligand binding and signalling is allosterically modulated by adenosine deaminase. Biochem. J. 2011, 435, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Gracia, E.; Cortes, A.; Meana, J.J.; Garcia-Sevilla, J.; Herhsfield, M.S.; Canela, E.I.; Mallol, J.; Lluis, C.; Franco, R.; Casado, V. Human adenosine deaminase as an allosteric modulator of human A(1) adenosine receptor: Abolishment of negative cooperativity for [H](R)-pia binding to the caudate nucleus. J. Neurochem. 2008, 107, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Fornai, M.; Awwad, O.; Giustarini, G.; Pellegrini, C.; Tuccori, M.; Caputi, V.; Qesari, M.; Castagliuolo, I.; Brun, P.; et al. Role of the A(2B) receptor-adenosine deaminase complex in colonic dysmotility associated with bowel inflammation in rats. Br. J. Pharmacol. 2014, 171, 1314–1329. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Canet, J.; Gracia, E.; Lluis, C.; Mallol, J.; Canela, E.I.; Cortes, A.; Casado, V. Molecular Evidence of Adenosine Deaminase Linking Adenosine A2A Receptor and CD26 Proteins. Front. Pharmacol. 2018, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Havre, P.A.; Abe, M.; Urasaki, Y.; Ohnuma, K.; Morimoto, C.; Dang, N.H. The role of CD26/dipeptidyl peptidase IV in cancer. Front. Biosci. 2008, 13, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.S.; Wakade, A.R. Quantitative analysis of similarities and differences in neurotoxicities caused by adenosine and 2′-deoxyadenosine in sympathetic neurons. J. Neurochem. 1996, 67, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Wakade, A.R.; Guo, X.; Palmer, K.C.; Kulkarni, J.S.; Przywara, D.A.; Wakade, T.D. 2′-deoxyadenosine induces apoptosis in rat chromaffin cells. J. Neurochem. 1996, 67, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gil, M.; Tozzi, M.G.; Allegrini, S.; Folcarelli, S.; Della Sala, G.; Voccoli, V.; Colombaioni, L.; Camici, M. Novel metabolic aspects related to adenosine deaminase inhibition in a human astrocytoma cell line. Neurochem. Int. 2012, 60, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gil, M.; Tozzi, M.G.; Varani, S.; Della Verde, L.; Petrotto, E.; Balestri, F.; Colombaioni, L.; Camici, M. The combination of adenosine deaminase inhibition and deoxyadenosine induces apoptosis in a human astrocytoma cell line. Neurochem. Int. 2015, 80, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gil, M.; Tozzi, M.G.; Balestri, F.; Colombaioni, L.; Camici, M. Mitochondrial Damage and Apoptosis Induced by Adenosine Deaminase Inhibition and Deoxyadenosine in Human Neuroblastoma Cell Lines. J. Cell. Biochem. 2016, 117, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.K.; Higashimori, H.; Tolman, M.; Yang, Y. Suppression of adenosine 2a receptor (A2aR)-mediated adenosine signaling improves disease phenotypes in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2015, 267, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, R.P.; Spector, T.; Parks, R.E. Tight-Binding Inhibitors. 4. Inhibition of Adenosine Deaminases by Various Inhibitors. Biochem. Pharmacol. 1977, 26, 359–367. [Google Scholar] [CrossRef]
- Dohner, H.; Ho, A.D.; Thaler, J.; Stryckmans, P.; Sonneveld, P.; de Witte, T.; Lechner, K.; Lauria, F.; Bodewadt-Radzun, S.; Suciu, S.; et al. Pentostatin in prolymphocytic leukemia: Phase II trial of the European Organization for Research and Treatment of Cancer Leukemia Cooperative Study Group. J. Natl. Cancer Inst. 1993, 85, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.R.; Goodrich, A.; Park, K.; Waselenko, J.K.; Lucas, M.; Reese, A.; Diehl, L.F.; Grever, M.R.; Byrd, J.C.; Flinn, I.W. A phase I/II study examining pentostatin, chlorambucil, and theophylline in patients with relapsed chronic lymphocytic leukemia and non-Hodgkin’s lymphoma. Ann. Hematol. 2006, 85, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Kay, N.E.; LaPlant, B.R.; Pettinger, A.M.; Call, T.G.; Leis, J.F.; Ding, W.; Parikh, S.A.; Conte, M.J.; Bowen, D.A.; Shanafelt, T.D. Cumulative experience and long term follow-up of pentostatin-based chemoimmunotherapy trials for patients with chronic lymphocytic leukemia. Expert Rev. Hematol. 2018, 11, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Rossi, D.; Motta, M.; Quaresmini, G.; Rossi, M.; Coscia, M.; Anastasia, A.; Rossini, F.; Cortelezzi, A.; Nador, G.; et al. A phase II multi-center trial of pentostatin plus cyclophosphamide with ofatumumab in older previously untreated chronic lymphocytic leukemia patients. Haematologica 2015, 100, e501–e504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, S.W., III; Hoffee, P.A. Adenosine deaminase from deoxycoformycin-sensitive and -resistant rat hepatoma cells. Purification and characterization. J. Biol. Chem. 1982, 257, 14239–14244. [Google Scholar] [PubMed]
- Camici, M.; Turriani, M.; Tozzi, M.G.; Turchi, G.; Cos, J.; Alemany, C.; Miralles, A.; Noe, V.; Ciudad, C.J. Purine enzyme profile in human colon-carcinoma cell lines and differential sensitivity to deoxycoformycin and 2′-deoxyadenosine in combination. Int. J. Cancer 1995, 62, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Bemi, V.; Tazzni, N.; Banditelli, S.; Giorgelli, F.; Pesi, R.; Turchi, G.; Mattana, A.; Sgarrella, F.; Tozzi, M.G.; Camici, M. Deoxyadenosine metabolism in a human colon-carcinoma cell line (LoVo) in relation to its cytotoxic effect in combination with deoxycoformycin. Int. J. Cancer 1998, 75, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Giannecchini, M.; D’Innocenzo, B.; Pesi, R.; Sgarrella, F.; Iorio, M.; Collecchi, P.; Tozzi, M.G.; Camici, M. 2′-Deoxyadenosine causes apoptotic cell death in a human colon carcinoma cell line. J. Biochem. Mol. Toxicol. 2003, 17, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Kutryb-Zajac, B.; Koszalka, P.; Mierzejewska, P.; Bulinska, A.; Zabielska, M.A.; Brodzik, K.; Skrzypkowska, A.; Zelazek, L.; Pelikant-Malecka, I.; Slominska, E.M.; et al. Adenosine deaminase inhibition suppresses progression of 4T1 murine breast cancer by adenosine receptor-dependent mechanisms. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Micheli, V.; Camici, M.; Tozzi, M.G.; Ipata, P.L.; Sestini, S.; Bertelli, M.; Pompucci, G. Neurological disorders of purine and pyrimidine metabolism. Curr. Top. Med. Chem. 2011, 11, 923–947. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, S.I.; Long, K.D.; Rosse, R.B.; Mastropaolo, J.; Eller, J. Hypothesized deficiency of guanine-based purines may contribute to abnormalities of neurodevelopment, neuromodulation, and neurotransmission in Lesch-Nyhan syndrome. Clin. Neuropharmacol. 2005, 28, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Picot, I.; Mockel, L.; Potier, M.C.; Dauphinot, L.; Shirley, T.L.; Torero-Ibad, R.; Fuchs, J.; Jinnah, H.A. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: Implications for Lesch-Nyhan disease pathogenesis. Hum. Mol. Genet. 2009, 18, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Zametkin, A.J.; Matochik, J.A.; Pascualvaca, D.; Jons, P.H.; Hardy, K.; Hankerson, J.G.; Doudet, D.J.; Cohen, R.M. Presynaptic dopaminergic deficits in Lesch-Nyhan disease. N. Engl. J. Med. 1996, 334, 1568–1572. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.F.; Harris, J.C.; Naidu, S.; Yokoi, F.; Marenco, S.; Dannals, R.F.; Ravert, H.T.; Yaster, M.; Evans, A.; Rousset, O.; et al. Dopamine transporters are markedly reduced in Lesch-Nyhan disease in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 5539–5543. [Google Scholar] [CrossRef] [PubMed]
- Schretlen, D.J.; Varvaris, M.; Vannorsdall, T.D.; Gordon, B.; Harris, J.C.; Jinnah, H.A. Brain white matter volume abnormalities in Lesch-Nyhan disease and its variants. Neurology 2015, 84, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.J.; Prior, C.; Garcia, M.G.; Puig, J.G. A review of the implication of hypoxanthine excess in the physiopathology of Lesch-Nyhan disease. Nucleosides Nucleotides Nucleic Acids 2016, 35, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Pelled, D.; Sperling, O.; Zoref-Shani, E. Abnormal purine and pyrimidine nucleotide content in primary astroglia cultures from hypoxanthine-guanine phosphoribosyltransferase-deficient transgenic mice. J. Neurochem. 1999, 72, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Zoref-Shani, E.; Bromberg, Y.; Brosh, S.; Sidi, Y.; Sperling, O. Characterization of the alterations in purine nucleotide metabolism in hypoxanthine-guanine phosphoribosyltransferase-deficient rat neuroma cell line. J. Neurochem. 1993, 61, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Shirley, T.L.; Lewers, J.C.; Egami, K.; Majumdar, A.; Kelly, M.; Ceballos-Picot, I.; Seidman, M.M.; Jinnah, H.A. A human neuronal tissue culture model for Lesch-Nyhan disease. J. Neurochem. 2007, 101, 841–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Sutcliffe, D.; Zhao, H.; Huang, X.; Schretlen, D.J.; Benkovic, S.; Jinnah, H.A. Clinical severity in Lesch-Nyhan disease: The role of residual enzyme and compensatory pathways. Mol. Genet. Metab. 2015, 114, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.H.; Stacey, N.C.; Connolly, G.P. Hypoxanthine impairs morphogenesis and enhances proliferation of a neuroblastoma model of Lesch Nyhan syndrome. J. Neurosci. Res. 2001, 63, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Spector, S. Identification of Inosine and Hypoxanthine as Endogenous Ligands for the Brain Benzodiazepine-Binding Sites. Proc. Natl. Acad. Sci. USA 1979, 76, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Anderson, L.T.; Reuben, R.; Dancis, J. Self-mutilation in Lesch-Nyhan disease is caused by dopaminergic denervation. Lancet 1985, 1, 338–339. [Google Scholar] [CrossRef]
- Torres, R.J.; Puig, J.G. Hypoxanthine deregulates genes involved in early neuronal development. Implications in Lesch-Nyhan disease pathogenesis. J. Inherit. Metab. Dis. 2015, 38, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.G.; Puig, J.G.; Torres, R.J. Adenosine, dopamine and serotonin receptors imbalance in lymphocytes of Lesch-Nyhan patients. J. Inherit. Metab. Dis. 2012, 35, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Frizzo, M.E.; Antunes Soares, F.A.; Dall’Onder, L.P.; Lara, D.R.; Swanson, R.A.; Souza, D.O. Extracellular conversion of guanine-based purines to guanosine specifically enhances astrocyte glutamate uptake. Brain Res. 2003, 972, 84–89. [Google Scholar] [CrossRef]
- Brosh, S.; Sperling, O.; Dantziger, E.; Sidi, Y. Metabolism of guanine and guanine nucleotides in primary rat neuronal cultures. J. Neurochem. 1992, 58, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.J.; Deantonio, I.; Prior, C.; Puig, J.G. Adenosine transport in peripheral blood lymphocytes from Lesch-Nyhan patients. Biochem. J. 2004, 377 Pt 3, 733–739. [Google Scholar] [CrossRef]
- Prior, C.; Torres, R.J.; Puig, J.G. Hypoxanthine effect on equilibrative and concentrative adenosine transport in human lymphocytes: Implications in the phatogenesis of Lesch-Nyhan syndrome. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; Gidday, J.M. Effect of dipyridamole on cerebral extracellular adenosine level in vivo. J. Cereb. Blood Flow Metab. 1990, 10, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Phillis, J.W.; O’Regan, M.H.; Walter, G.A. Effects of two nucleoside transport inhibitors, dipyridamole and soluflazine, on purine release from the rat cerebral cortex. Brain Res. 1989, 481, 309–316. [Google Scholar] [CrossRef]
- Newby, A.C. How does dipyridamole elevate extracellular adenosine concentration? Predictions from a three-compartment model of adenosine formation and inactivation. Biochem. J. 1986, 237, 845–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasibetti, H.; Pierozan, P.; Rodrigues, A.F.; Manfredini, V.; Wyse, A.T.S. Hypoxanthine Intrastriatal Administration Alters Neuroinflammatory Profile and Redox Status in Striatum of Infant and Young Adult Rats. Mol. Neurobiol. 2017, 54, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Biasibetti-Brendler, H.; Schmitz, F.; Pierozan, P.; Zanotto, B.S.; Prezzi, C.A.; de Andrade, R.B.; Wannmacher, C.M.D.; Wyse, A.T.S. Hypoxanthine Induces Neuroenergetic Impairment and Cell Death in Striatum of Young Adult Wistar Rats. Mol. Neurobiol. 2018, 55, 4098–4106. [Google Scholar] [CrossRef] [PubMed]
- Sidi, Y.; Mitchell, B.S. Z-nucleotide accumulation in erythrocytes from Lesch-Nyhan patients. J. Clin. Investig. 1985, 76, 2416–2419. [Google Scholar] [CrossRef] [PubMed]
- Marie, S.; Heron, B.; Bitoun, P.; Timmerman, T.; Van Den Berghe, G.; Vincent, M.F. AICA-ribosiduria: A novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 2004, 74, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gil, M.; Pesi, R.; Perna, S.; Allegrini, S.; Giannecchini, M.; Camici, M.; Tozzi, M.G. 5′-aminoimidazole-4-carboxamide riboside induces apoptosis in human neuroblastoma cells. Neuroscience 2003, 117, 811–820. [Google Scholar] [CrossRef]
- Lopez, J.M. Is ZMP the toxic metabolite in Lesch-Nyhan disease? Med. Hypotheses 2008, 71, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Ishii, K.; Nanmoku, T.; Isobe, K.; Kawakami, Y.; Takekoshi, K. 5-Aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside stimulates tyrosine hydroxylase activity and catecholamine secretion by activation of AMP-activated protein kinase in PC12 cells. J. Neuroendocrinol. 2007, 19, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Zhao, H.; Pugh, R.J.; Pedley, A.M.; French, J.; Jones, S.A.; Zhuang, X.; Jinnah, H.; Huang, T.J.; Benkovic, S.J. Purinosome formation as a function of the cell cycle. Proc. Natl. Acad. Sci. USA 2015, 112, 1368–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, C.S.; Jinnah, H.A.; Shirley, T.L.; Nyhan, W.L.; Seifert, R. Altered membrane NTPase activity in Lesch-Nyhan disease fibroblasts: Comparison with HPRT knockout mice and HPRT-deficient cell lines. J. Neurochem. 2005, 93, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, V.; Pinto, C.S.; Seifert, R. Complex changes in ecto-nucleoside 5′-triphosphate diphosphohydrolase expression in hypoxanthine phosphoribosyl transferase deficiency. Neurosci. Lett. 2007, 420, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Erdorf, M.; von der Ohe, J.; Seifert, R. Impaired P2X and P2Y receptor-mediated signaling in HPRT-deficient B103 neuroblastoma cells. Neurosci. Lett. 2011, 504, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, L.; Kim, J.E.; Miyanohara, A.; Kang, T.H.; Friedmann, T. Purinergic signaling in human pluripotent stem cells is regulated by the housekeeping gene encoding hypoxanthine guanine phosphoribosyltransferase. Proc. Natl. Acad. Sci. USA 2012, 109, 3377–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Park, Y.; Bader, J.S.; Friedmann, T. The housekeeping gene hypoxanthine guanine phosphoribosyltransferase (HPRT) regulates multiple developmental and metabolic pathways of murine embryonic stem cell neuronal differentiation. PLoS ONE 2013, 8, e74967. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.; Fagan, B.M.; Magness, S.T.; Hutton, S.; Taranova, O.; Hayashi, S.; McMahon, A.; Rao, M.; Pevny, L. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci. 2004, 26, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Guibinga, G.H.; Barron, N.; Pandori, W. Striatal neurodevelopment is dysregulated in purine metabolism deficiency and impacts DARPP-32, BDNF/TrkB expression and signaling: New insights on the molecular and cellular basis of Lesch-Nyhan Syndrome. PLoS ONE 2014, 9, e96575. [Google Scholar] [CrossRef] [PubMed]
- Guibinga, G.H.; Hrustanovic, G.; Bouic, K.; Jinnah, H.A.; Friedmann, T. MicroRNA-mediated dysregulation of neural developmental genes in HPRT deficiency: Clues for Lesch-Nyhan disease? Hum. Mol. Genet. 2012, 21, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Friedmann, T. Alzheimer’s disease shares gene expression aberrations with purinergic dysregulation of HPRT deficiency (Lesch-Nyhan disease). Neurosci. Lett. 2015, 590, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Camici, M.; Tozzi, M.G.; Allegrini, S.; Del Corso, A.; Sanfilippo, O.; Daidone, M.G.; De Marco, C.; Ipata, P.L. Purine salvage enzyme activities in normal and neoplastic human tissues. Cancer Biochem. Biophys. 1990, 11, 201–209. [Google Scholar] [PubMed]
- Sanfilippo, O.; Camici, M.; Tozzi, M.G.; Turriani, M.; Faranda, A.; Ipata, P.L.; Silvestrini, R. Relationship between the levels of purine salvage pathway enzymes and clinical/biological aggressiveness of human colon carcinoma. Cancer Biochem. Biophys. 1994, 14, 57–66. [Google Scholar] [PubMed]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.H.; Felsted, A.M.; Ence, Z.E.; Piccolo, S.R.; Robison, R.A.; O’Neill, K.L. Elevated Expression of Hypoxanthine Guanine Phosphoribosyltransferase within Malignant Tissue. Cancer Clin. Oncol. 2017, 6, 19–34. [Google Scholar] [CrossRef]
- Townsend, M.H.; Anderson, M.D.; Weagel, E.G.; Velazquez, E.J.; Weber, K.S.; Robison, R.A.; O’Neill, K.L. Non-small-cell lung cancer cell lines A549 and NCI-H460 express hypoxanthine guanine phosphoribosyltransferase on the plasma membrane. OncoTargets Ther. 2017, 10, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.H.; Robison, R.A.; O’Neill, K.L. A review of HPRT and its emerging role in cancer. Med. Oncol. 2018, 35, 89. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Nishino, T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 1995, 9, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Bortolotti, M.; Polito, L.; Bolognesi, A. The role of xanthine oxidoreductase and uric acid in metabolic syndrome. Biochim. Biophys. Acta 2018, 1864, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorbach, C.; Scriven, A.; Capecchi, M.R. The housekeeping gene xanthine oxidoreductase is necessary for milk fat droplet enveloping and secretion: Gene sharing in the lactating mammary gland. Genes Dev. 2002, 16, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.K.; Naeher, T.M.; Broome, J.E.; Boedeker, E.C. Role of host xanthine oxidase in infection due to enteropathogenic and Shiga-toxigenic Escherichia coli. Infect. Immun. 2013, 81, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Simeunovic Ostojic, M.; Maas, J. Anorexia nervosa and uric acid beyond gout: An idea worth researching. Int. J. Eat Disord. 2018, 51, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Sevanian, A.; Muakkassah-Kelly, S.F.; Hochstein, P. Uric acid-iron ion complexes. A new aspect of the antioxidant functions of uric acid. Biochem. J. 1986, 235, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Lario, B.; Macarron-Vicente, J. Is there anything good in uric acid? QJM 2011, 104, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, E.K.; Boison, D.; Schwarzschild, M.A.; Kochanek, P.M. Purines: Forgotten mediators in traumatic brain injury. J. Neurochem. 2016, 137, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Mount, D.B.; Kwon, C.Y.; Zandi-Nejad, K. Renal urate transport. Rheum. Dis. Clin. 2006, 32, 313–331. [Google Scholar] [CrossRef] [PubMed]
- Hisatome, I.; Tsuboi, M.; Shigemasa, C. Renal hypouricemia. Nihon Rinsho 1996, 54, 3337–3342. [Google Scholar] [PubMed]
- Sharaf El Din, U.A.A.; Salem, M.M.; Abdulazim, D.O. Uric acid in the pathogenesis of metabolic, renal, and cardiovascular diseases: A review. J. Adv. Res. 2017, 8, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.; Chen, Y.D.I.; Hollenbeck, C.B.; Reaven, G.M. Relationship between Resistance to Insulin-Mediated Glucose-Uptake, Urinary Uric-Acid Clearance, and Plasma Uric-Acid Concentration. J. Am. Med. Assoc. 1991, 266, 3008–3011. [Google Scholar] [CrossRef]
- Kang, D.H.; Chen, W. Uric acid and chronic kidney disease: New understanding of an old problem. Semin. Nephrol. 2011, 31, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Perheentupa, J.; Raivio, K. Fructose-induced hyperuricaemia. Lancet 1967, 2, 528–531. [Google Scholar] [CrossRef]
- Bjorge, T.; Lukanova, A.; Jonsson, H.; Tretli, S.; Ulmer, H.; Manjer, J.; Stocks, T.; Selmer, R.; Nagel, G.; Almquist, M.; et al. Metabolic syndrome and breast cancer in the me-can (metabolic syndrome and cancer) project. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Haffner, S.M.; Baillargeon, J. Adiposity, the metabolic syndrome, and breast cancer in African-American and white American women. Endocr. Rev. 2007, 28, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E. Metabolic syndrome, hyperinsulinemia, and colon cancer: A review. Am. J. Clin. Nutr. 2007, 86, s836–s842. [Google Scholar] [CrossRef] [PubMed]
- Fini, M.A.; Elias, A.; Johnson, R.J.; Wright, R.M. Contribution of uric acid to cancer risk, recurrence, and mortality. Clin. Transl. Med. 2012, 1, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzali, M.; Kanbay, M.; Segal, M.S.; Shafiu, M.; Jalal, D.; Feig, D.I.; Johnson, R.J. Uric acid and hypertension: Cause or effect? Curr. Rheumatol. Rep. 2010, 12, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Sofaer, J.A.; Emery, A.E. Genes for super-intelligence? J. Med. Genet. 1981, 18, 410–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orowan, E. The origin of man. Nature 1955, 175, 683–684. [Google Scholar] [CrossRef] [PubMed]
- Cervini, C.; Zampa, A.M. Uric-Acid and Intelligence. Ann. Rheum. Dis. 1982, 41, 435. [Google Scholar] [CrossRef]
- Inouye, E.; Park, K.S.; Asaka, A. Blood uric acid level and IQ: A study in twin families. Acta Genet. Med. Gemellol. 1984, 33, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.S.; Hooper, D.C. The role of uric acid in protection against peroxynitrite-mediated pathology. Med. Hypotheses 2001, 56, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Kori, M.; Aydin, B.; Unal, S.; Arga, K.Y.; Kazan, D. Metabolic Biomarkers and Neurodegeneration: A Pathway Enrichment Analysis of Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. OMICS 2016, 20, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- de Lau, L.M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Serum uric acid levels and the risk of Parkinson disease. Ann. Neurol. 2005, 58, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Lolekha, P.; Wongwan, P.; Kulkantrakorn, K. Association between serum uric acid and motor subtypes of Parkinson’s disease. J. Clin. Neurosci. 2015, 22, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ng, S.Y.; Chia, N.S.; Acharyya, S.; Setiawan, F.; Lu, Z.H.; Ng, E.; Tay, K.Y.; Au, W.L.; Tan, E.K.; et al. Serum uric acid level and its association with motor subtypes and non-motor symptoms in early Parkinson’s disease: PALS study. Parkinsonism Relat. Disord. 2018, 55, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, L.; Teng, W.; Huang, K.; Shang, X. Correlation of fatigue during the acute stage of stroke with serum uric acid and glucose levels, depression, and disability. Eur. Neurol. 2014, 72, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; Bright, A.T.; Alaynick, W.A.; Wang, L.; Baxter, A.; Nathan, N.; Anderson, W.; Gordon, E. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E5472–E5480. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Hao, D.L.; Wu, B.N.; Mao, L.L.; Zhang, J. Uric acid demonstrates neuroprotective effect on Parkinson’s disease mice through Nrf2-ARE signaling pathway. Biochem. Biophys. Res. Commun. 2017, 493, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Keizman, D.; Ish-Shalom, M.; Berliner, S.; Maimon, N.; Vered, Y.; Artamonov, I.; Tsehori, J.; Nefussy, B.; Drory, V.E. Low uric acid levels in serum of patients with ALS: Further evidence for oxidative stress? J. Neurol. Sci. 2009, 285, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kato, M.; Kusano, T.; Nishino, T. New Strategy That Delays Progression of Amyotrophic Lateral Sclerosis in G1H-G93A Transgenic Mice: Oral Administration of Xanthine Oxidoreductase Inhibitors That Are Not Substrates for the Purine Salvage Pathway. J. Neuropathol. Exp. Neurol. 2016, 75, 1124–1144. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.; Obach, V.; Cervera, A.; Revilla, M.; Deulofeu, R.; Aponte, J.H. Prognostic significance of uric acid serum concentration in patients with acute ischemic stroke. Stroke 2002, 33, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.; Amaro, S.; Castellanos, M.; Segura, T.; Arenillas, J.; Marti-Fabregas, J.; Gallego, J.; Krupinski, J.; Gomis, M.; Canovas, D.; et al. Safety and efficacy of uric acid in patients with acute stroke (URICO-ICTUS): A randomised, double-blind phase 2b/3 trial. Lancet Neurol. 2014, 13, 453–460. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Macklin, E.A.; Ascherio, A.; Parkinson Study Group SURE-PD Investigators. Urate and neuroprotection trials. Lancet Neurol. 2014, 13, 758. [Google Scholar] [CrossRef]
- Dachir, S.; Shabashov, D.; Trembovler, V.; Alexandrovich, A.G.; Benowitz, L.I.; Shohami, E. Inosine improves functional recovery after experimental traumatic brain injury. Brain Res. 2014, 1555, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Zai, L.; Liang, P.; Schaffling, C.; Ahlborn, D.; Benowitz, L.I. Inosine enhances axon sprouting and motor recovery after spinal cord injury. PLoS ONE 2013, 8, e81948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Chen, G.J.; Harvey, B.K.; Bickford, P.C.; Wang, Y. Inosine reduces ischemic brain injury in rats. Stroke 2005, 36, 654–659. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Gil, M.; Camici, M.; Allegrini, S.; Pesi, R.; Petrotto, E.; Tozzi, M.G. Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors. Int. J. Mol. Sci. 2018, 19, 3598. https://doi.org/10.3390/ijms19113598
Garcia-Gil M, Camici M, Allegrini S, Pesi R, Petrotto E, Tozzi MG. Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors. International Journal of Molecular Sciences. 2018; 19(11):3598. https://doi.org/10.3390/ijms19113598
Chicago/Turabian StyleGarcia-Gil, Mercedes, Marcella Camici, Simone Allegrini, Rossana Pesi, Edoardo Petrotto, and Maria Grazia Tozzi. 2018. "Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors" International Journal of Molecular Sciences 19, no. 11: 3598. https://doi.org/10.3390/ijms19113598
APA StyleGarcia-Gil, M., Camici, M., Allegrini, S., Pesi, R., Petrotto, E., & Tozzi, M. G. (2018). Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors. International Journal of Molecular Sciences, 19(11), 3598. https://doi.org/10.3390/ijms19113598