Loss of Response Gene to Complement 32 (RGC-32) in Diabetic Mouse Retina Is Involved in Retinopathy Development


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
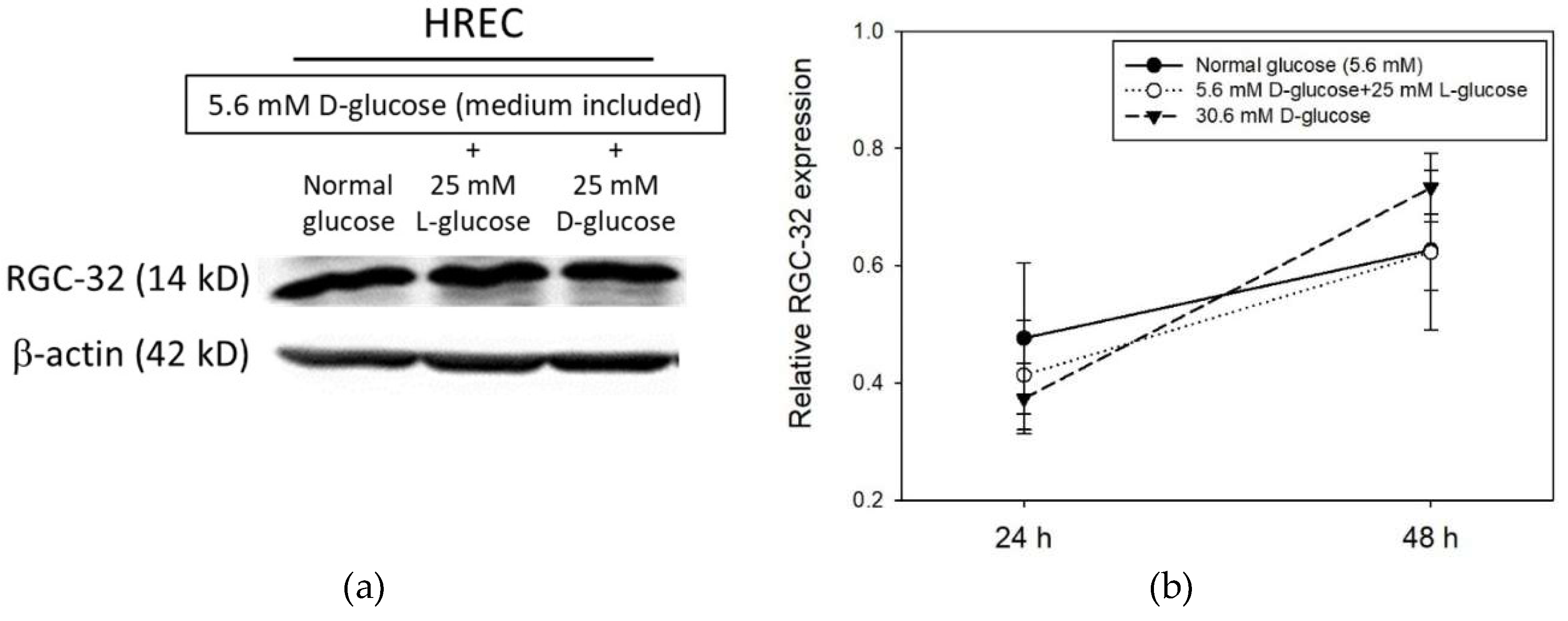
2.1. RGC-32 Expression in Human Retinal Cells under Hyperglycemic Condition
2.2. Histopathology in T2D Mouse Retina
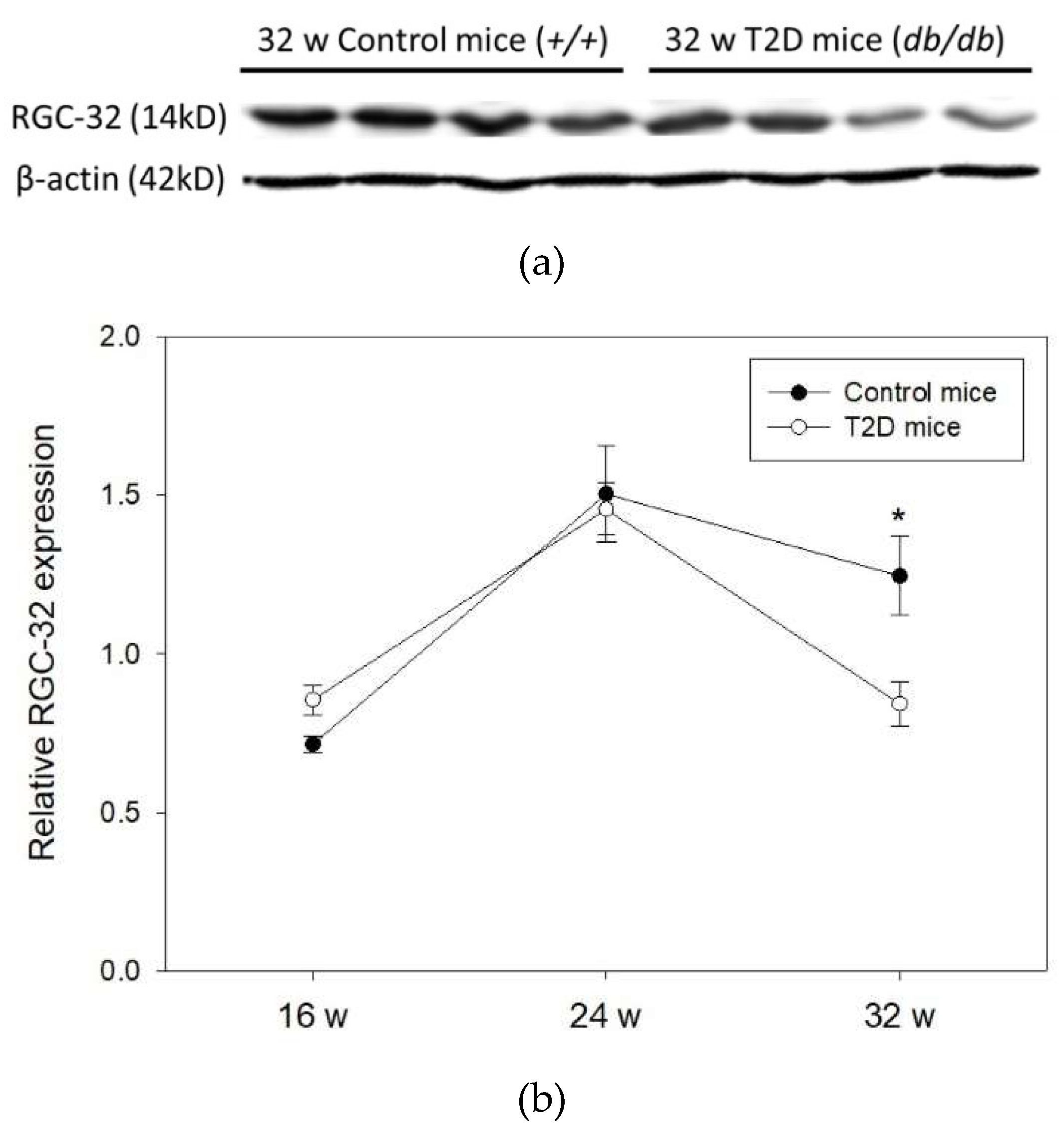
2.3. Time-Dependent Change in RGC-32 Expression in T2D Mouse Retina
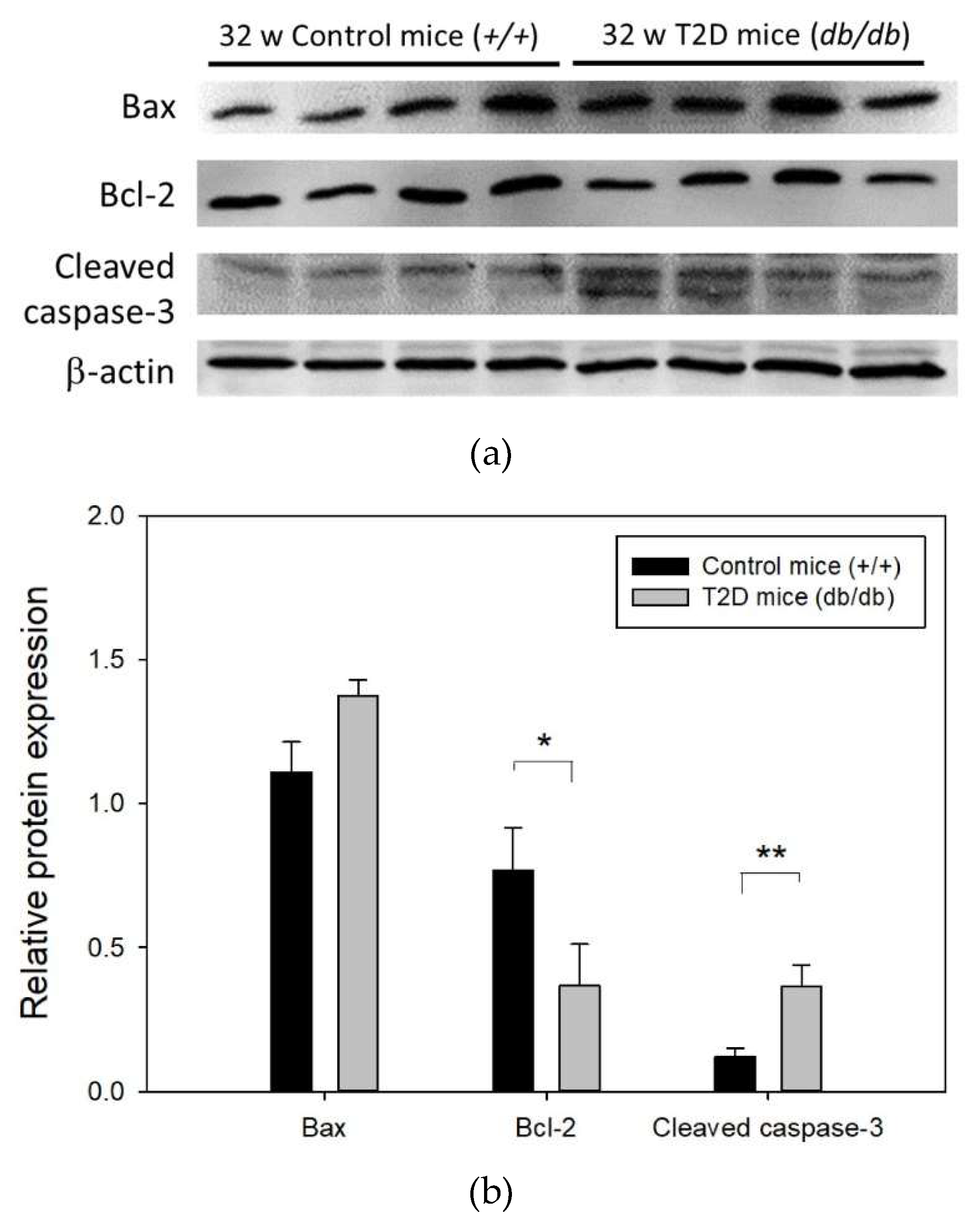
2.4. Increased Apoptosis of Retinal Cells in T2D Mice at 32 Weeks of Age
3. Discussion:
4. Materials and Methods
4.1. Cell Culture
4.2. Type 2 Diabetes (T2D) Mouse Model
4.3. Retinal Histopathology
4.4. Western Blot
4.5. Immunohistochemistry
4.6. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Bax | BCL2-associated X |
| Bcl-2 | B-cell lymphoma 2 |
| DR | diabetic retinopathy |
| GCL | ganglion cell layer |
| HREC | human retinal microvascular endothelial cells |
| INL | inner nuclear layer |
| IPL | inner plexiform layer |
| ONL | outer nuclear layers |
| RGC-32 | response gene to complements 32 |
| RPE | retinal pigment epithelium |
| T2D | type 2 diabetes |
| VEGF | vascular endothelial growth factor |
References
- Moss, S.E.; Klein, R.; Klein, B.E. The 14-year incidence of visual loss in a diabetic population. Ophthalmology 1998, 105, 998–1003. [Google Scholar] [CrossRef]
- Taylor, H.R.; Keeffe, J.E. World blindness: A 21st century perspective. Br. J. Ophthalmol. 2001, 85, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Cikamatana, L.; Mitchell, P.; Rochtchina, E.; Foran, S.; Wang, J.J. Five-year incidence and progression of diabetic retinopathy in a defined older population: The blue mountains eye study. Eye 2007, 21, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Jerneld, B.; Algvere, P. Relationship of duration and onset of diabetes to prevalence of diabetic retinopathy. Am. J. Ophthalmol. 1986, 102, 431–437. [Google Scholar] [CrossRef]
- Leske, M.C.; Wu, S.Y.; Hennis, A.; Hyman, L.; Nemesure, B.; Yang, L.; Schachat, A.P. Hyperglycemia, blood pressure, and the 9-year incidence of diabetic retinopathy: The barbados eye studies. Ophthalmology 2005, 112, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Looker, H.C.; Krakoff, J.; Knowler, W.C.; Bennett, P.H.; Klein, R.; Hanson, R.L. Longitudinal studies of incidence and progression of diabetic retinopathy assessed by retinal photography in pima indians. Diabetes Care 2003, 26, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Hsu, Y.M.; Lin, Y.J.; Huang, Y.C.; Chen, C.J.; Lin, W.D.; Liao, W.L.; Chen, Y.T.; Lin, W.Y.; Liu, Y.H.; et al. Current concepts regarding developmental mechanisms in diabetic retinopathy in taiwan. Biomedicine (Taipei) 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Badea, T.; Niculescu, F.; Soane, L.; Fosbrink, M.; Sorana, H.; Rus, V.; Shin, M.L.; Rus, H. RGC-32 increases p34CDC2 kinase activity and entry of aortic smooth muscle cells into s-phase. J. Biol. Chem. 2002, 277, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Fosbrink, M.; Cudrici, C.; Tegla, C.A.; Soloviova, K.; Ito, T.; Vlaicu, S.; Rus, V.; Niculescu, F.; Rus, H. Response gene to complement 32 is required for C5b-9 induced cell cycle activation in endothelial cells. Exp. Mol. Pathol. 2009, 86, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Luo, Z.; Huang, W.; Lu, Q.; Wilcox, C.S.; Jose, P.A.; Chen, S. Response gene to complement 32, a novel regulator for transforming growth factor-beta-induced smooth muscle differentiation of neural crest cells. J. Biol. Chem. 2007, 282, 10133–10137. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.N.; Shi, N.; Xie, W.B.; Guo, X.; Chen, S.Y. Response gene to complement 32 promotes vascular lesion formation through stimulation of smooth muscle cell proliferation and migration. Arterioscler. Thromb. Vasc. Biol. 2011, 31, e19–e26. [Google Scholar] [CrossRef] [PubMed]
- Badea, T.C.; Niculescu, F.I.; Soane, L.; Shin, M.L.; Rus, H. Molecular cloning and characterization of RGC-32, a novel gene induced by complement activation in oligodendrocytes. J. Biol. Chem. 1998, 273, 26977–26981. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shang, C.; Zhao, J.; Han, Y.; Liu, J.; Chen, K.; Shi, W. Knockdown of response gene to complement 32 (RGC32) induces apoptosis and inhibits cell growth, migration, and invasion in human lung cancer cells. Mol. Cell. Biochem. 2014, 394, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Fosbrink, M.; Cudrici, C.; Niculescu, F.; Badea, T.C.; David, S.; Shamsuddin, A.; Shin, M.L.; Rus, H. Overexpression of RGC-32 in colon cancer and other tumors. Exp. Mol. Pathol. 2005, 78, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Lee, J.Y.; Lee, S.M.; Choi, J.E.; Cho, S.; Park, J.Y. Promoter methylation of the RGC32 gene in nonsmall cell lung cancer. Cancer 2011, 117, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Vlaicu, S.I.; Tegla, C.A.; Cudrici, C.D.; Fosbrink, M.; Nguyen, V.; Azimzadeh, P.; Rus, V.; Chen, H.; Mircea, P.A.; Shamsuddin, A.; et al. Epigenetic modifications induced by RGC-32 in colon cancer. Exp. Mol. Pathol. 2010, 88, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaicu, S.I.; Cudrici, C.; Ito, T.; Fosbrink, M.; Tegla, C.A.; Rus, V.; Mircea, P.A.; Rus, H. Role of response gene to complement 32 in diseases. Arch. Immunol. Ther. Exp. 2008, 56, 115–122. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Jin, Y.; Guo, H.; Foo, S.Y.; Cully, B.L.; Wu, J.; Zeng, H.; Rosenzweig, A.; Li, J. Response gene to complement 32, a novel hypoxia-regulated angiogenic inhibitor. Circulation 2009, 120, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.B.; Luan, J.N.; Ye, J.; Chen, S.Y. RGC32 deficiency protects against high-fat diet-induced obesity and insulin resistance in mice. J. Endocrinol. 2015, 224, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Philbrick, M.J.; An, X.; Xu, M.; Wu, J. Response gene to complement 32 (RGC-32) in endothelial cells is induced by glucose and helpful to maintain glucose homeostasis. Int. J. Clin. Exp. Med. 2014, 7, 2541–2549. [Google Scholar] [PubMed]
- Cui, X.B.; Luan, J.N.; Chen, S.Y. Rgc-32 deficiency protects against hepatic steatosis by reducing lipogenesis. J. Biol. Chem. 2015, 290, 20387–20395. [Google Scholar] [CrossRef] [PubMed]
- Feenstra, D.J.; Yego, E.C.; Mohr, S. Modes of retinal cell death in diabetic retinopathy. J. Clin. Exp. Ophthalmol. 2013, 4, 298. [Google Scholar] [PubMed]
- Kern, T.S.; Berkowitz, B.A. Photoreceptors in diabetic retinopathy. J. Diabetes Investig. 2015, 6, 371–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanov, P.; Corraliza, L.; Villena, J.A.; Carvalho, A.R.; Garcia-Arumi, J.; Ramos, D.; Ruberte, J.; Simo, R.; Hernandez, C. The db/db mouse: A useful model for the study of diabetic retinal neurodegeneration. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, Y.; Jiang, Y.; Willard, L.; Ortiz, E.; Wark, L.; Medeiros, D.; Lin, D. Dietary wolfberry ameliorates retinal structure abnormalities in db/db mice at the early stage of diabetes. Exp. Biol. Med. 2011, 236, 1051–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Lu, Q.; Chen, W.; Li, J.; Li, C.; Zheng, Z. Vitamin D3 protects against diabetic retinopathy by inhibiting high-glucose-induced activation of the ROS/TXNIP/NLRP3 inflammasome pathway. J. Diabetes Res. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, W.-L.; Lin, J.-M.; Liu, S.-P.; Chen, S.-Y.; Lin, H.-J.; Wang, Y.-H.; Lei, Y.-J.; Huang, Y.-C.; Tsai, F.-J. Loss of Response Gene to Complement 32 (RGC-32) in Diabetic Mouse Retina Is Involved in Retinopathy Development. Int. J. Mol. Sci. 2018, 19, 3629. https://doi.org/10.3390/ijms19113629
Liao W-L, Lin J-M, Liu S-P, Chen S-Y, Lin H-J, Wang Y-H, Lei Y-J, Huang Y-C, Tsai F-J. Loss of Response Gene to Complement 32 (RGC-32) in Diabetic Mouse Retina Is Involved in Retinopathy Development. International Journal of Molecular Sciences. 2018; 19(11):3629. https://doi.org/10.3390/ijms19113629
Chicago/Turabian StyleLiao, Wen-Ling, Jane-Ming Lin, Shih-Ping Liu, Shih-Yin Chen, Hui-Ju Lin, Yeh-Han Wang, Yu-Jie Lei, Yu-Chuen Huang, and Fuu-Jen Tsai. 2018. "Loss of Response Gene to Complement 32 (RGC-32) in Diabetic Mouse Retina Is Involved in Retinopathy Development" International Journal of Molecular Sciences 19, no. 11: 3629. https://doi.org/10.3390/ijms19113629
APA StyleLiao, W. -L., Lin, J. -M., Liu, S. -P., Chen, S. -Y., Lin, H. -J., Wang, Y. -H., Lei, Y. -J., Huang, Y. -C., & Tsai, F. -J. (2018). Loss of Response Gene to Complement 32 (RGC-32) in Diabetic Mouse Retina Is Involved in Retinopathy Development. International Journal of Molecular Sciences, 19(11), 3629. https://doi.org/10.3390/ijms19113629