Genome-Wide Identification of Flowering-Time Genes in Brassica Species and Reveals a Correlation between Selective Pressure and Expression Patterns of Vernalization-Pathway Genes in Brassica napus

 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Identification of Flowering-Time Genes in B. rapa, B. oleracea, and B. napus
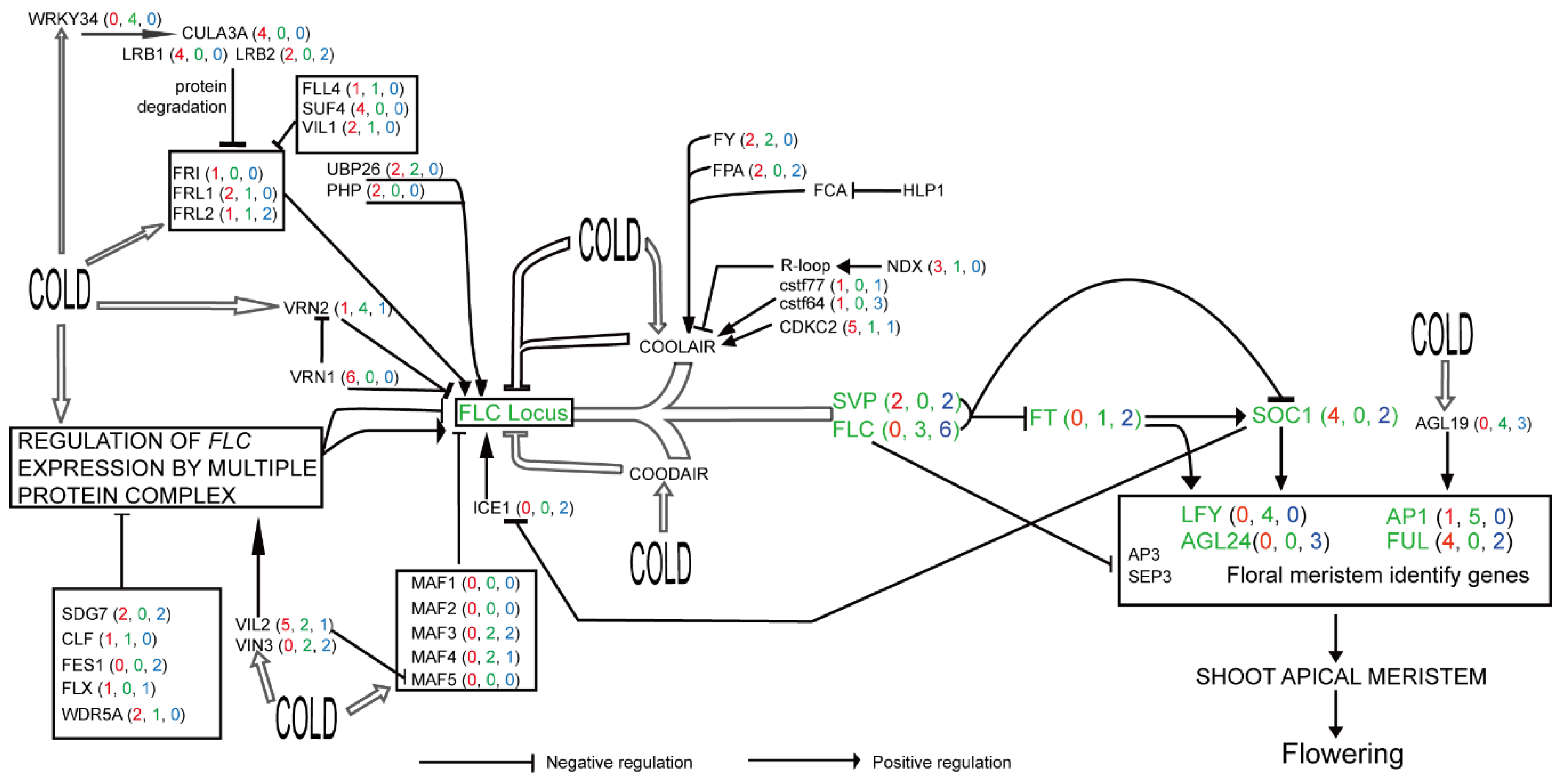
2.2. Partition of Flowering-Time Genes to Different Pathways
2.3. Chromosomal Location Analysis
2.4. Gene Expansion Analysis
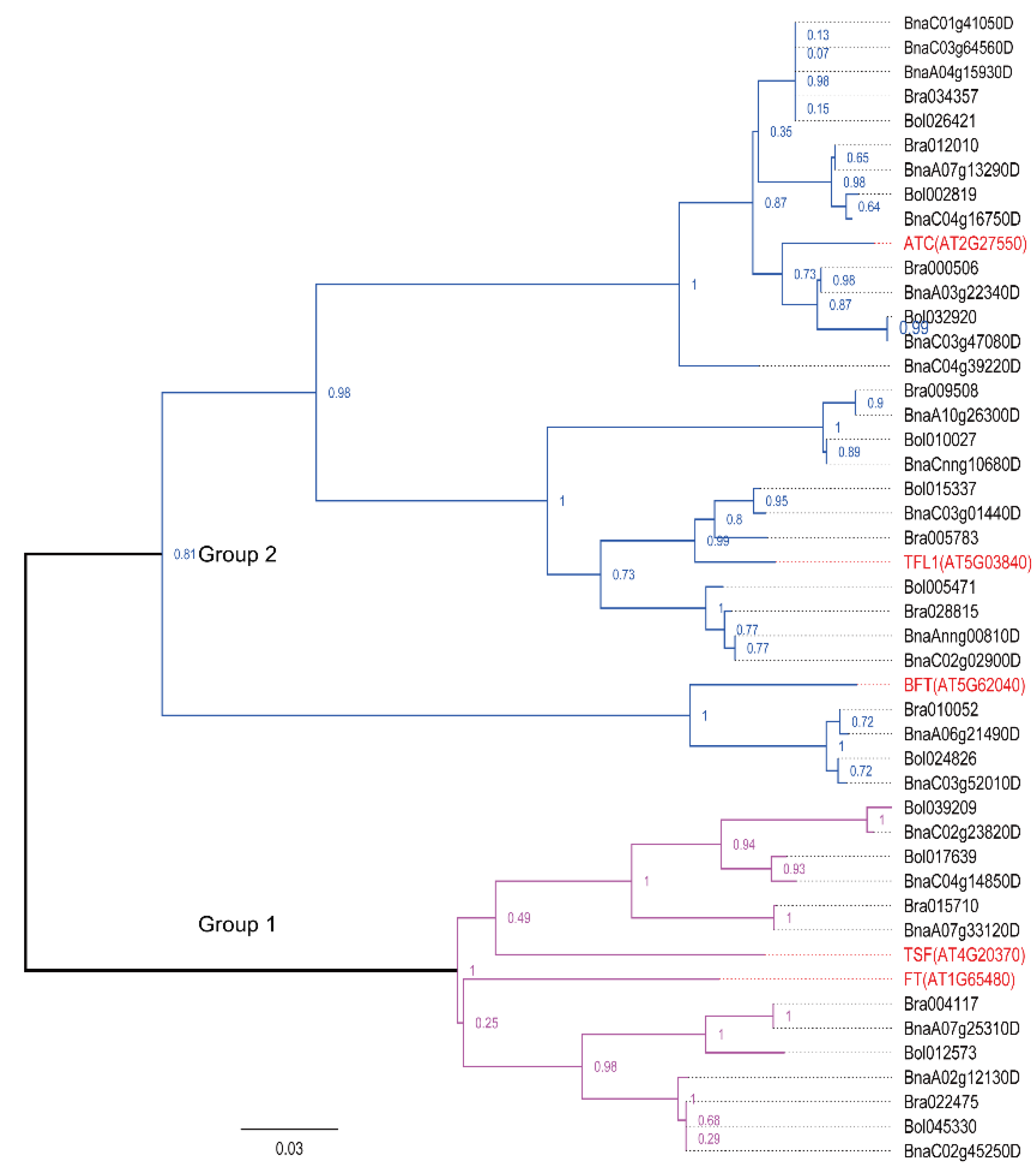
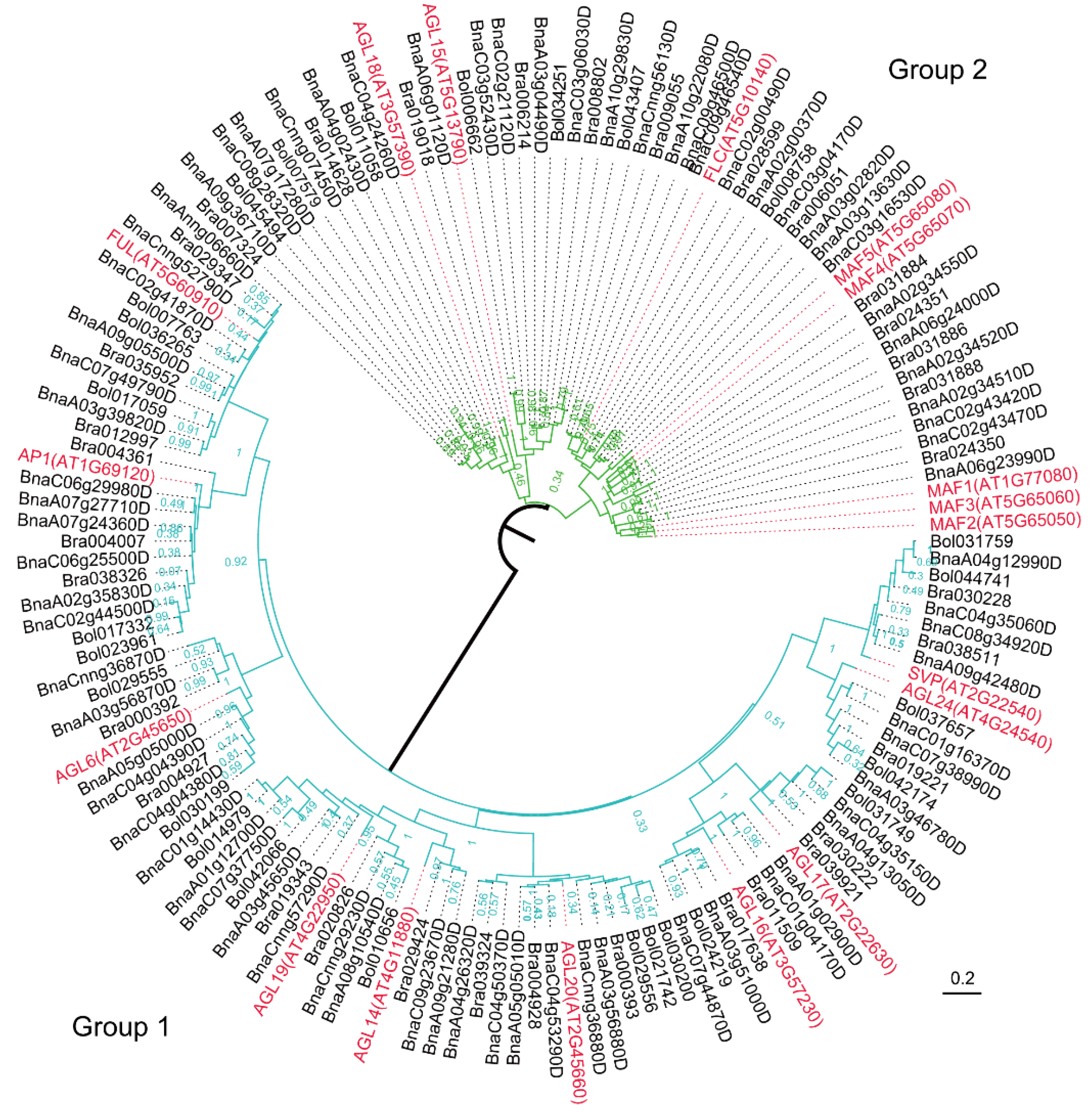
2.5. Phylogenetic Analysis of Two Flowering-Time Gene Families
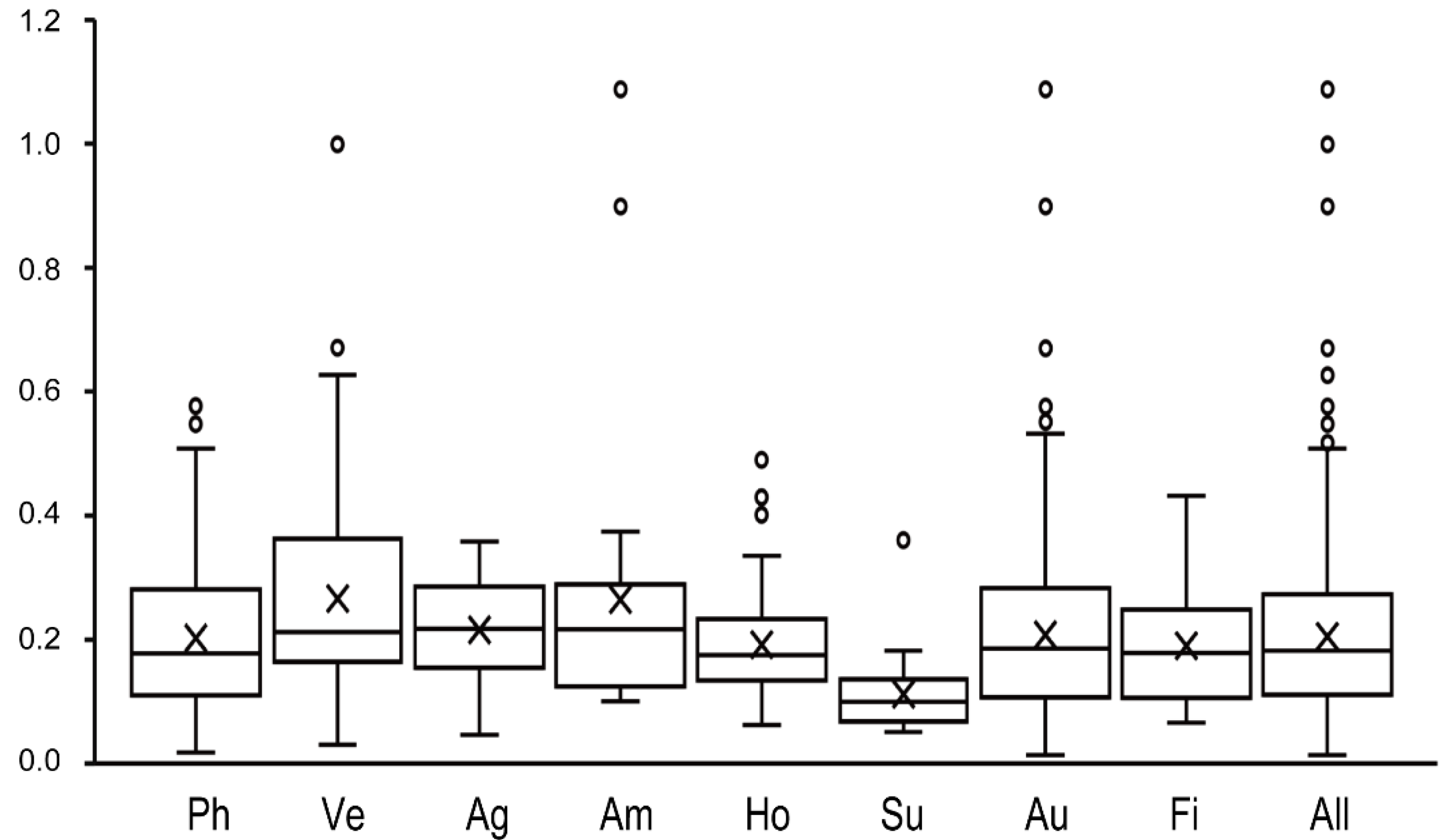
2.6. Selective Pressure Calculations of Flowering-Time Genes
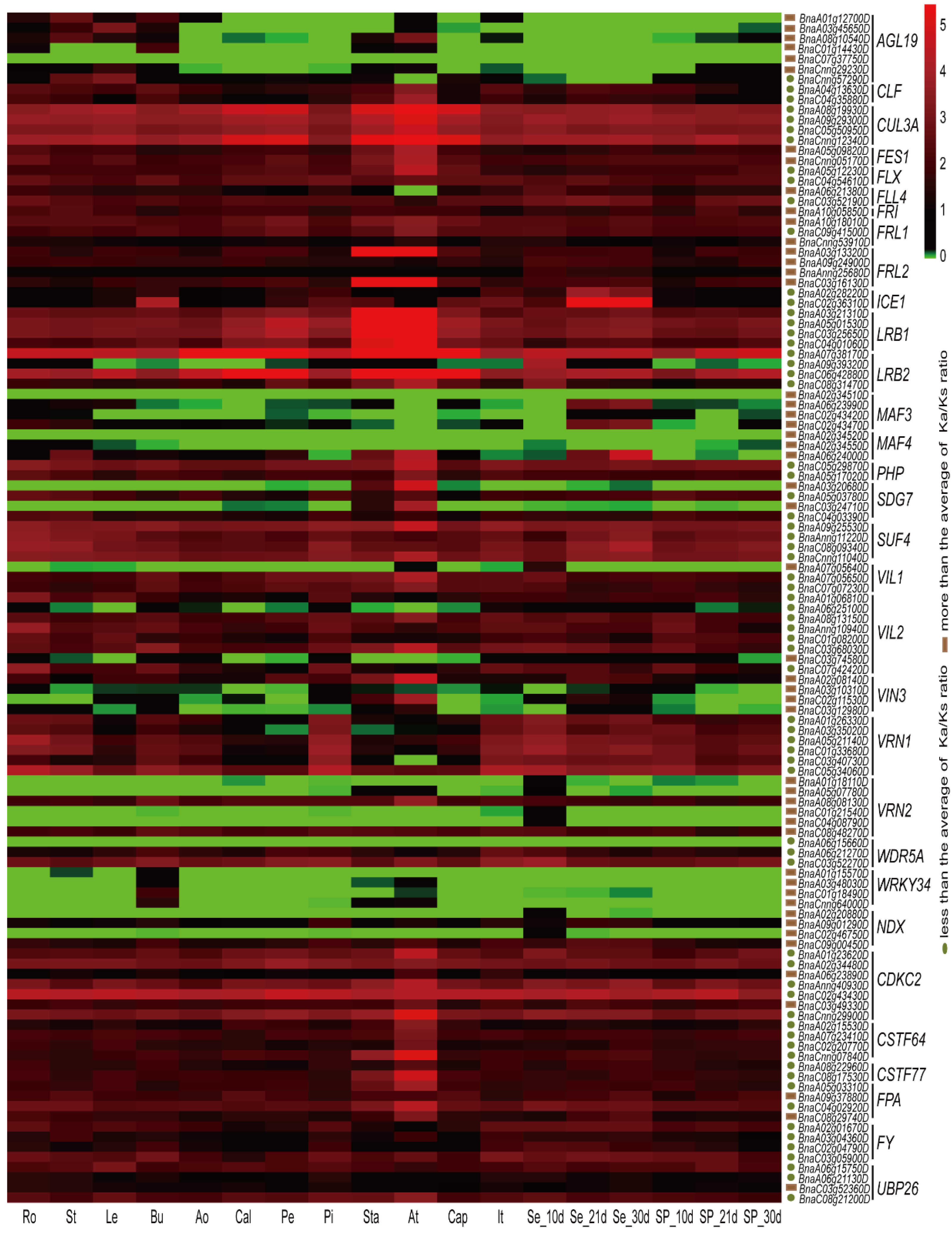
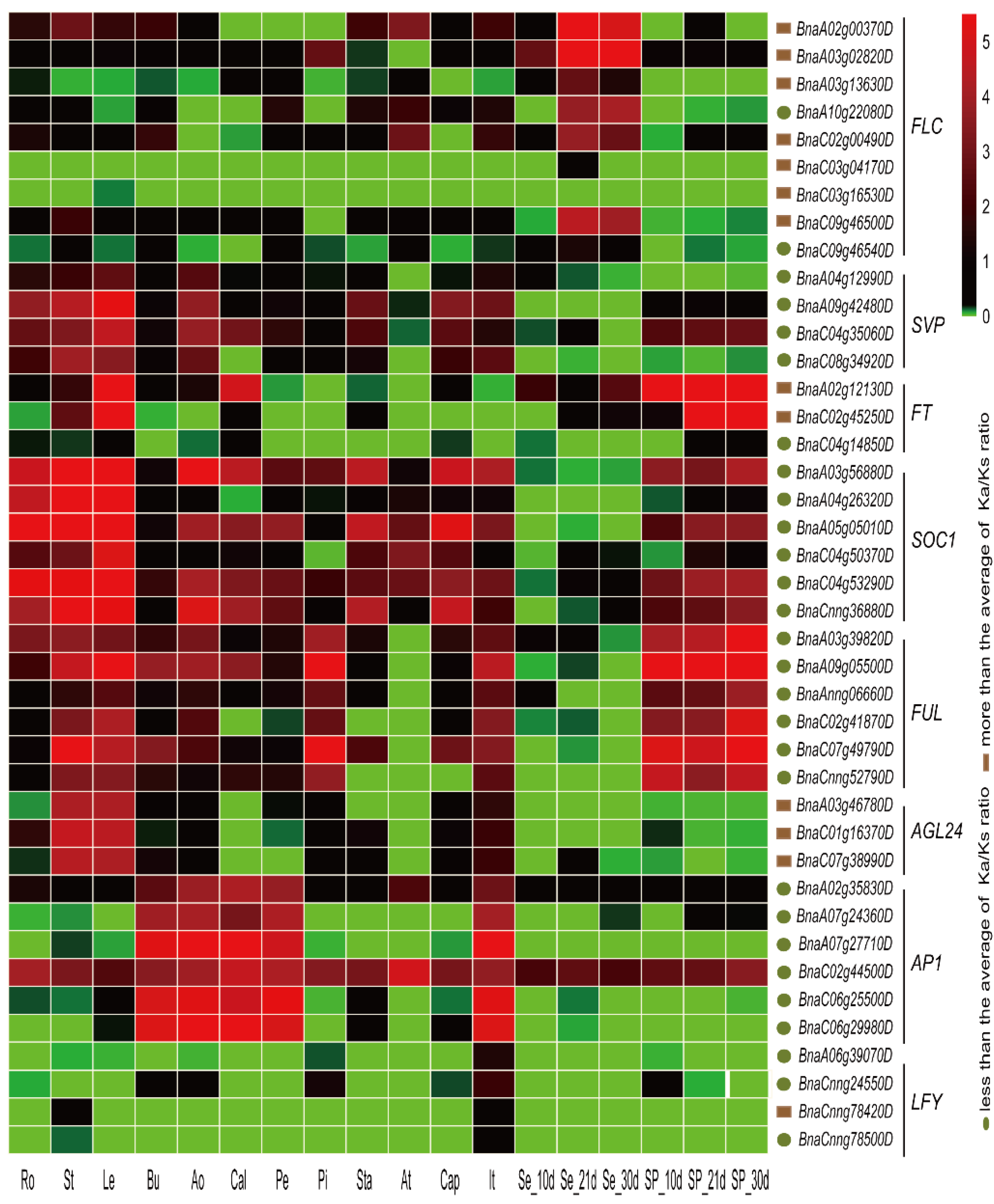
2.7. Expression Patterns of Vernalization-Pathway Genes
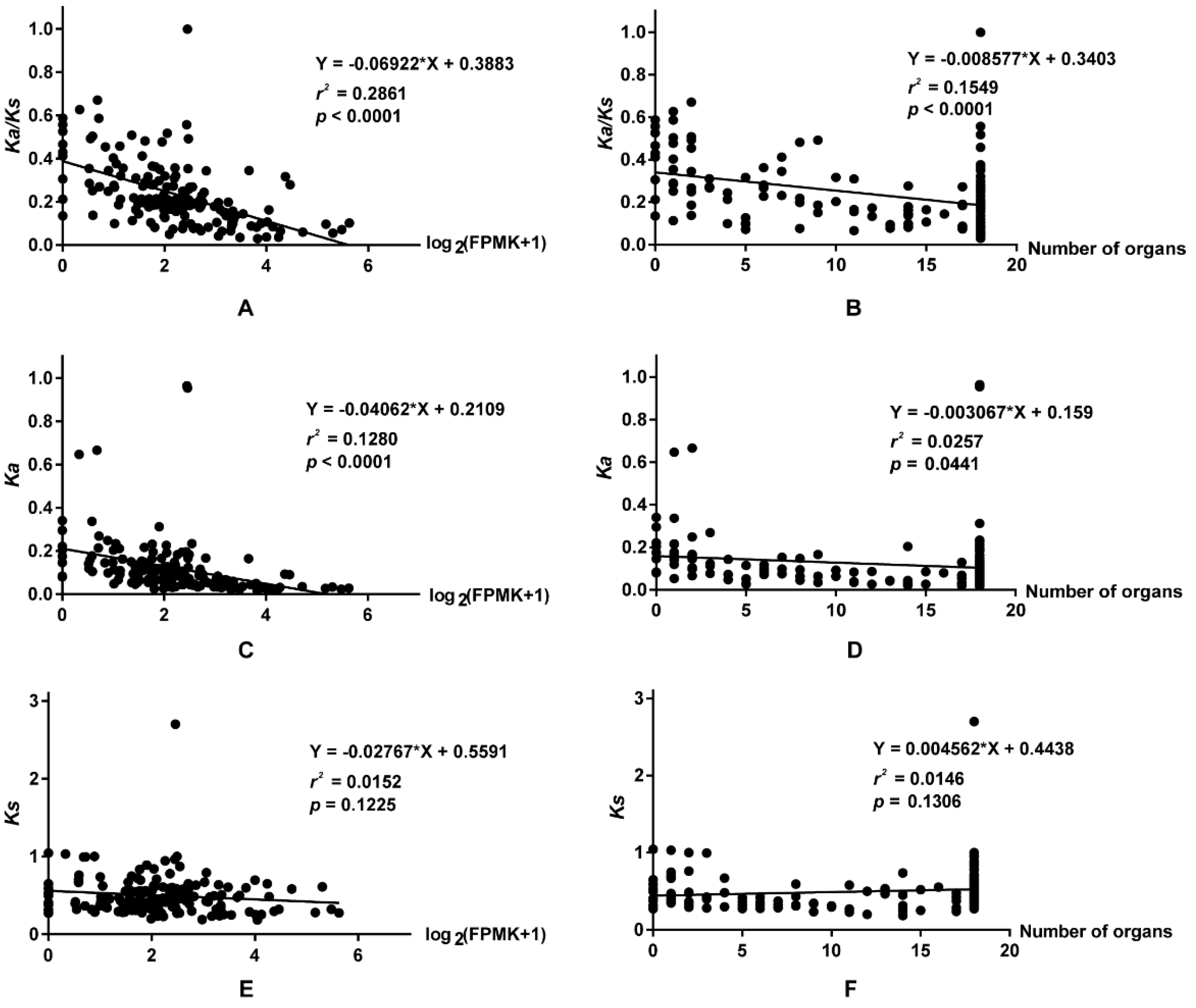
2.8. The relationship between Ka/Ks Ratios and Expression Patterns
3. Discussion
4. Materials and Methods
4.1. Data Resources
4.2. Identification of Flowering-Time Genes in Brassica species
4.3. Chromosomal Location and Gene Expansion Analysis
- C1: gene number in flowering-time pathway A in B. rapa or B. oleracea
- C2: gene number in flowering-time pathway A in B. napus
- N1: total coding gene number in B. rapa or B. oleracea
- N2: total coding gene number in B. napus
4.4. Phylogenetic Analysis
4.5. Ka/Ks Calculation of Flowering-Time Genes in B. napus
4.6. Expression Profiles of Vernalization-Pathway Genes in B. napus
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Andres, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Blackman, B.K.; Rasmussen, D.A.; Strasburg, J.L.; Raduski, A.R.; Burke, J.M.; Knapp, S.J.; Michaels, S.D.; Rieseberg, L.H. Contributions of flowering time genes to sunflower domestication and improvement. Genetics 2011, 187, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Flowers, J.M.; Hanzawa, Y.; Hall, M.C.; Moore, R.C.; Purugganan, M.D. Population genomics of the Arabidopsis thaliana flowering time gene network. Mol. Biol. Evol. 2009, 26, 2475–2486. [Google Scholar] [CrossRef] [PubMed]
- Purugganan, M.D.; Fuller, D.Q. The nature of selection during plant domestication. Nature 2009, 457, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.A.; Bailey, P.C.; Laurie, D.A. Comparative Genomics of Flowering Time Pathways Using Brachypodium distachyon as a Model for the Temperate Grasses. PLoS ONE 2010, 5, e10065. [Google Scholar] [CrossRef] [PubMed]
- Jack, T. Molecular and genetic mechanisms of floral control. Plant Cell 2004, 16 (Suppl. 1), S1–S17. [Google Scholar] [CrossRef] [PubMed]
- Lukens, L.; Zou, F.; Lydiate, D.; Parkin, I.; Osborn, T. Comparison of a Brassica oleracea genetic map with the genome of Arabidopsis thaliana. Genetics 2003, 164, 359–372. [Google Scholar] [PubMed]
- Tan, F.C.; Swain, S.M. Genetics of flower initiation and development in annual and perennial plants. Physiol. Plant. 2006, 128, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Weigel, D.; Nilsson, O. A developmental switch sufficient for flower initiation in diverse Plant. Nature 1995, 377, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, M.A.; Weigel, D. Integration of floral inductive signals in Arabidopsis. Nature 2000, 404, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Bouché, F.; Lobet, G.; Tocquin, P.; Périlleux, C. FLOR-ID: An interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016, 44, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Harmer, S.L. The circadian system in higher plants. Annu. Rev. Plant Biol. 2009, 60, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T. Arabidopsis circadian clock and photoperiodism: Time to think about location. Curr. Opin. Plant Biol. 2010, 13, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Kay, S.A. Photoperiodic control of flowering: Not only by coincidence. Trends Plant Sci. 2006, 11, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Putterill, J.; Robson, F.; Lee, K.; Simon, R.; Coupland, G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factor. Cell 1995, 95, 847–857. [Google Scholar] [CrossRef]
- Samach, A.; Onouchi, H.; Gold, S.E.; Ditta, G.S.; Schwarz-Sommer, Z.; Yanofsky, M.F.; Coupland, G. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Sciences 2000, 288, 1613–1616. [Google Scholar] [CrossRef]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell. Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef] [PubMed]
- Blümel, M.; Dally, N.; Jung, C. Flowering time regulation in crops-what did we learn from Arabidopsis? Curr. Opin. Biotechnol. 2015, 32, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Michaels, S.D.; Amasino, R.M. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, C.C.; Burn, J.E.; Perez, P.P.; Metzger, J.; Edwards, J.A.; Peacock, W.J.; Dennis, E.S. The FLF MADS box gene: A repressor of flowering in Arabidopsis regulated by vernalization and methylation. Plant Cell 1999, 11, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Colasanti, J.; Coneva, V. Mechanisms of floral induction in grasses: Something borrowed, something new. Plant Physiol. 2009, 149, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Grover, C.E.; Gallagher, J.P.; Wendel, J.F. Candidate gene identification of flowering time genes in cotton. Plant Genome 2015, 8, 1–13. [Google Scholar] [CrossRef]
- Turck, F.; Fornara, F.; Coupland, G. Regulation and identity of florigen: FLOWERING LOCUS T moves center stage. Annu. Rev. Plant Biol. 2008, 59, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Qiu, Y.; Cheng, F.; Chen, X.H.; Zhang, Z.H.; Wang, H.P.; Song, J.P.; Duan, M.M.; Yang, H.H.; Li, X.X. Genome-wide identification, characterization, and evolutionary analysis of flowering genes in radish (Raphanus sativus L.). BMC Genomics 2017, 18, 981. [Google Scholar] [CrossRef] [PubMed]
- Wilczek, A.M.; Roe, J.L.; Knapp, M.C.; Cooper, M.D.; Lopez-Gallego, C.; Martin, L.J.; Muir, C.D.; Sim, S.; Walker, A.; Anderson, J. Effects of genetic perturbation on seasonal life history plasticity. Science 2009, 323, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.A.; Lister, C.; Soumpourou, E.; Zhang, Y.E.; Howell, E.C.; Teakle, G.; Dean, C. Functional alleles of the flowering time regulator FRIGIDA in the Brassica oleracea genome. BMC Plant Biol. 2012, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Schiessl, S.V.; Huettel, B.; Kuehn, D.; Reinhardt, R.; Snowdon, R.J. Flowering Time Gene Variation in Brassica Species Shows Evolutionary Principles. Front. Plant Sci. 2017, 8, 1742. [Google Scholar] [CrossRef] [PubMed]
- Schranz, M.E.; Quijada, P.; Sung, S.B.; Lukens, L.; Amasino, R.; Osborn, T.C. Characterization and effects of the replicated flowering time gene FLC in Brassica rapa. Genetics 2002, 162, 1457–1468. [Google Scholar] [PubMed]
- Zhang, X.M.; Meng, L.; Liu, B.; Hu, Y.Y.; Cheng, F.; Liang, J.L.; Aarts, M.G.M.; Wang, X.W.; Wu, J. A transposon insertion in FLOWERING LOCUS T is associated with delayed flowering in Brassica rapa. Plant Sci. 2015, 241, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Wenkel, S.; Turck, F.; Singer, K.; Gissot, L.; Le Gourrierec, J.; Samach, A.; Coupland, G. Constans and the ccaat box binding complex share a functionally important domain and interact to regulate flowering of Arabidopsis. Plant Cell 2006, 18, 2971–2984. [Google Scholar] [CrossRef] [PubMed]
- Parcy, F. Flowering: A time for integration. Int. J. Dev. Biol. 2005, 49, 585. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Nagaharu, U.; Nagaharu, N. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 1935, 7, 389–452. [Google Scholar]
- Wang, X.W.; Wang, H.Z.; Wang, J.; Sun, R.F.; Wu, J.; Liu, S.Y.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Liu, Y.M.; Yang, X.H.; Tong, C.B.; Edwards, D.; Parkin, I.A.P.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef] [PubMed]
- Karlgren, A.; Gyllenstrand, N.; Kallmanetal, T. Evolution of the PEBP gene family in plants: Functional diversification in seed plant evolution. Plant Physiol. 2011, 156, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Kawahigashi, H.; Ando, T.; Tonooka, T.; Handa, H. Molecular and functional characterization of PEBP genes in barley reveal the diversification of their roles in flowering. Plant Physiol. 2009, 149, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Fadina, O.A.; Pankin, A.A.; Khavkin, A.A. Molecular characterization of the flowering time gene FRIGIDA in Brassica genomes A and C. Russ. J. Plant Physiol. 2013, 60, 279–289. [Google Scholar] [CrossRef]
- Razi, H.; Howell, E.C.; Newbury, H.J.; Kearsey, M.J. Does sequence polymorphism of FLC paralogues underlie flowering time QTL in Brassica oleracea. Theor. Appl. Genet. 2008, 116, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, B.; Hemming, M.N.; Dennis, E.S.; Peacock, W.J. The molecular basis of vernalization-induced flowering in cereals. Trends Plant Sci. 2007, 12, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wu, J.; Wang, X.W. Genome triplication drove the diversification of Brassica plants. Hortic. Res. 2014, 1, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.Y.; Hu, Z.Q.; Yan, C.R. Genome-Wide Comparative Analysis of Flowering-Related Genes in Arabidopsis, Wheat, and Barley. Int. J. Plant Genomics 2015, 17, 874361. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.I.; Wang, J.G.; Poon, S.Y.; Su, C.L.; Wang, S.S.; Chiou, T.J. Differential regulation of FLOWERING LOCUS C expression by vernalization in cabbage and Arabidopsis. Plant Physiol. 2005, 137, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Sakamoto, K.; Kikuchi, R.; Saito, A.; Togashi, E.; Kuginuki, Y.; Matsumoto, S.; Hirai, M. Mapping and characterization of FLC homologs and QTL analysis of flowering time in Brassica oleracea. Theor. Appl. Genet. 2007, 114, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Tadege, M.; Sheldon, C.C.; Helliwell, C.A.; Stoutjesdijk, P.; Dennis, E.S.; Peacock, W.J. Control of flowering time by FLC orthologues in Brassica napus. Plant J. 2001, 28, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Weinholdt, C.; Jedrusik, N.; Molina, C.; Zou, J.; Große, I.; Schiess, S.; Jung, C.; Emrani, N. Whole-transcriptome analysis reveals genetic factors underlying flowering time regulation in rapeseed (Brassica napus L.). Plant Cell Environ. 2018, 41, 1935–1947. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hu, K.; Zhang, Z.Q.; Guan, C.Y.; Chen, S.; Hua, W.; Li, J.N.; Wen, J.; Yi, B.; Shen, J.X.; et al. Genome-wide association study reveals the geneticarchitectureof floweringtimeinrapeseed (Brassica napus L.). DNA Res. 2016, 23, 43–52. [Google Scholar] [PubMed]
- Cockram, J.; Jones, H.; Leigh, F.J.; O’Sullivan, D.; Powell, W.; Laurie, D.A.; Greenland, A.J. Control of flowering time in temperate cereals: Genes, domestication, and sustainable productivity. J. Exp. Bot. 2007, 58, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Risk, J.M.; Laurie, R.E.; Macknight, R.C.; Day, C.L. FRIGIDA and related proteins have a conserved central domain and family specific N- and C-terminal regions that are functionally important. Plant Mol. Biol. 2010, 73, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L. Adaptive evolution after gene duplication. Trends Genet. 2002, 18, 433–434. [Google Scholar] [CrossRef]
- Kondrashov, F.A.; Rogozin, I.B.; Wolf, Y.I.; Koonin, E.V. Selection in the evolution of gene duplications. Genome Biol. 2002, 2, 81–89. [Google Scholar]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A. Decoupled evolution of coding region and mRNA expression patterns after gene duplication: Implications for the neutralist-selectionist debate. Proc. Natl. Acad. Sci. USA 2000, 97, 6579–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, D.A.; Raval, A.; Wilke, C.O. A single determinant dominates the rate of yeast protein evolution. Mol. Biol. Evol. 2006, 23, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Papp, B.; Hurst, L.D. Highly expressed genes in yeast evolve slowly. Genetics 2001, 158, 927–931. [Google Scholar] [PubMed]
- Subramanian, S.; Kumar, S. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics 2004, 168, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Duret, L.; Mouchiroud, D. Determinants of substitution rates in mammalian genes: Expression pattern affects selection intensity but not mutation rate. Mol. Biol. Evol. 2000, 17, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, W.H. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol. Biol. Evol. 2004 21, 236–239. [CrossRef]
- Guo, Y.; Liu, J.; Zhang, J.F.; Liu, S.Y.; Du, J.C. Selective modes determine evolutionary rates, gene compactness and expression patterns in Brassica. Plant J. 2017, 91, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gaut, B.S. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol. Biol. Evol. 2011, 28, 2359–2369. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H. Molecular Evolution, 1st ed.; Sinauer Associates Incorporated: Sunderland, MA, USA, 1997. [Google Scholar]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gaped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.H.; Yu, M.N.; Liu, M.; Zhang, R.; Sun, W.; Qian, M.C.; Duan, H.C.; Chang, W.; Ma, J.Q.; Qu, C.M.; et al. Genome-Wide Identification, Evolutionary and Expression Analyses of the GALACTINOL SYNTHASE Gene Family in Rapeseed and Tobacco. Int. J. Mol. Sci. 2017, 18, 2768. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. ClustalW and ClustalX version2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, K.; Glen, S.; Koichiro, T. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Batesian evolutionary analysis sample trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MRBAYES 3: Bayesian phylogenetic inference under mixed mode. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. CONSEL: For assessing the confidence of phylogenetic tree selection. Bioinformatics 2001, 17, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Li, W.H.; Wu, C.I.; Luo, C.C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 1985, 2, 150–174. [Google Scholar] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat. Protocol. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways | A. thaliana | B. oleracea | B. rapa | B. napus | |||
|---|---|---|---|---|---|---|---|
| Number of Genes | Ratio * | Number of Genes | Ratio * | Number of Genes | Ratio * | ||
| Ph | 90 (113) | 156 (193) | 1.71 | 153 (187) | 1.66 | 318 (390) | 3.45 |
| Ve | 26 (34) | 41 (57) | 1.68 | 46 (61) | 1.79 | 88 (117) | 3.44 |
| Ag | 2 (8) | 4 (13) | 1.63 | 4 (12) | 1.50 | 9 (25) | 3.13 |
| Am | 5 (10) | 6 (12) | 1.20 | 7 (13) | 1.30 | 16 (27) | 2.70 |
| Ho | 25 (25) | 47 (47) | 1.88 | 43 (43) | 1.72 | 99 (99) | 3.96 |
| Su | 9 (9) | 17 (17) | 1.89 | 15 (15) | 1.67 | 29 (29) | 3.22 |
| Au | 88 (113) | 163 (209) | 1.85 | 155 (198) | 1.75 | 325 (413) | 3.66 |
| Fi | 16 (16) | 33 (33) | 2.06 | 34 (34) | 2.13 | 72 (72) | 4.50 |
| Ph/Ag | 6 | 9 | - | 8 | - | 16 | - |
| Am/Au | 2 | 4 | - | 4 | - | 9 | - |
| Am/Ph | 2 | 2 | - | 2 | - | 4 | - |
| Am/Ve | 1 | 0 | - | 0 | - | 0 | - |
| Ph/Au | 15 | 26 | - | 24 | - | 50 | - |
| Ve/Au | 8 | 16 | - | 15 | - | 29 | - |
| Total | 295 | 524 | 1.78 | 510 | 1.729 | 1064 | 3.607 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Fan, Y.; Yu, J.; Chai, L.; Zhang, J.; Jiang, J.; Cui, C.; Zheng, B.; Jiang, L.; Lu, K. Genome-Wide Identification of Flowering-Time Genes in Brassica Species and Reveals a Correlation between Selective Pressure and Expression Patterns of Vernalization-Pathway Genes in Brassica napus. Int. J. Mol. Sci. 2018, 19, 3632. https://doi.org/10.3390/ijms19113632
Li H, Fan Y, Yu J, Chai L, Zhang J, Jiang J, Cui C, Zheng B, Jiang L, Lu K. Genome-Wide Identification of Flowering-Time Genes in Brassica Species and Reveals a Correlation between Selective Pressure and Expression Patterns of Vernalization-Pathway Genes in Brassica napus. International Journal of Molecular Sciences. 2018; 19(11):3632. https://doi.org/10.3390/ijms19113632
Chicago/Turabian StyleLi, Haojie, Yonghai Fan, Jingyin Yu, Liang Chai, Jingfang Zhang, Jun Jiang, Cheng Cui, Benchuan Zheng, Liangcai Jiang, and Kun Lu. 2018. "Genome-Wide Identification of Flowering-Time Genes in Brassica Species and Reveals a Correlation between Selective Pressure and Expression Patterns of Vernalization-Pathway Genes in Brassica napus" International Journal of Molecular Sciences 19, no. 11: 3632. https://doi.org/10.3390/ijms19113632
APA StyleLi, H., Fan, Y., Yu, J., Chai, L., Zhang, J., Jiang, J., Cui, C., Zheng, B., Jiang, L., & Lu, K. (2018). Genome-Wide Identification of Flowering-Time Genes in Brassica Species and Reveals a Correlation between Selective Pressure and Expression Patterns of Vernalization-Pathway Genes in Brassica napus. International Journal of Molecular Sciences, 19(11), 3632. https://doi.org/10.3390/ijms19113632