Is the Response of Tumours Dependent on the Dietary Input of Some Amino Acids or Ratios among Essential and Non-Essential Amino Acids? All That Glitters Is Not Gold

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Nutrition and the Risk of Cancer: The Puzzling Question of Insulin Resistance and Type-2 Diabetes. Are Amino Acid Plasma Patterns the Cause or Effect?
3. Essential Amino Acids and Cancer: What if the Substrates Provided by the Environment Change the Rules?
4. Reducing the EAA/NEAA Ratios Drives Cancer Cells to Apoptosis, Activating Autophagy and Inhibiting the Proteasome
5. Serine and Glycine: Guilty of Feeding Cancer or Innocent Intermediates of Metabolism?
6. Glutamine, Proline, and Ornithine—Entangled Relationship
7. Conclusions
Funding
Conflicts of Interest
References
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Vaitheesvaran, B.; Xu, J.; Yee, J.; Lu, Q.-Y.; Go, V.L.; Xia, O.; Lee, W.N. The Warburg effect: A balance of flux analysis. Metabolomics 2015, 11, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Elti, N.; Striebel, M.; Ulset, A.J.; Cross, W.F.; De Vilbiss, S.; Glibert, P.M.; Guo, L.; Hirst, A.G.; Hood, J.; Kominoski, J.S.; et al. Bridging food webs, ecosystem metabolism, and biogeochemistry using ecological stoichiometry theory. Front. Microbiol. 2017, 12, 1298. [Google Scholar] [CrossRef]
- Chinn, S.B.; Darr, O.A.; Peters, R.D.; Prince, M.E. The role of head and neck squamous cell carcinoma cancer stem cells in tumorigenesis, metastasis, and treatment failure. Front. Endocrin. 2012, 3, 90. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.D.; Delneri, D.; Oliver, S.G.; Rattray, M.; Bergman, C.M. Evolutionary Systems Biology of Amino Acid Biosynthetic Cost in Yeast. PLoS ONE 2010, 5, E11935. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.P.; Thompson, C.B. Cancer cell metabolism: The essential role of the non-essential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guan, J.Z.; Sun, Y.; Le, Z.; Zhang, P.; Yu, D.; Liu, Y. Insulin-like growth factor 1 receptor-mediated cell survival in hypoxia depends on the promotion of autophagy via suppression of the PI3K/Akt/mTOR signaling pathway. Mol. Med. Rep. 2017, 15, 2136–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhanwar-Uniyal, M.; Amin, A.G.; Cooper, J.B.; Das, K.; Schmidt, M.H.; Murali, R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv. Biol. Reg. 2017, 64, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Alves, T.; Zhao, X.; Cline, G.W.; Zhang, D.; Bhanot, S.; Samuel, V.T.; Kibbey, R.G.; Shulman, G.I. Arginino succinate synthetase regulates hepatic AMPK linking protein catabolism and ureagenesis to hepatic lipid metabolism. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef]
- Gerber, K. Targeting mTOR: Something old, something new. J. Natl. Cancer Inst. 2009, 101, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Glynne-Jones, R. Challenges behind proving efficacy of adjuvant chemotherapy after preoperative chemoradiation for rectal cancer. Lancet Oncol. 2017, 18, E354–E363. [Google Scholar] [CrossRef]
- Pulvers, J.N.; Marx, G. Factors associated with the development and severity of oxaliplatin-induced peripheral neuropathy: A systematic review. Asia-Pac. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Macarulla, T.; Fernández, T.; Gallardo, M.E.; Hernando, O.; López, A.M.; Hidalgo, M. Adjuvant treatment for pancreatic ductal carcinoma. Clin. Transl. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Saba, N.F.; Mody, M.D.; Tan, E.S.; Gill, H.S.; Rinaldo, A.; Takes, R.P.; Strojan, P.; Hartl, D.M.; Vermorken, J.B.; Haigentz, M., Jr.; et al. Toxicities of systemic agents in squamous cell carcinoma of the head and neck (SCCHN); A new perspective in the era of immunotherapy. Crit. Rev. Oncol. Hematol. 2017, 115, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Porta, C.A.M.; Zapperi, S.; Sethna, J.P. Senescent Cells in Growing Tumors: Population Dynamics and Cancer Stem Cells. PLoS Comput. Biol. 2012, 8, E1002316. [Google Scholar] [CrossRef] [PubMed]
- Sherman, C.D., Jr.; Morton, J.J.; Mider, G.B. Potential Sources of Tumor Nitrogen. Cancer Res. 1950, 10, 374–378. [Google Scholar] [PubMed]
- Maddocks, O.D.K.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013. [Google Scholar] [CrossRef] [PubMed]
- Bonfili, L.; Cecarini, V.; Cuccioloni, M.; Angeletti, M.; Flati, V.; Corsetti, G.; Pasini, E.; Dioguardi, F.S.; Eleuteri, A.M. Essential amino acid mixtures drive cancer cells to apoptosis through proteasome inhibition and autophagy activation. FEBS J. 2017, 284, 1726–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, K.D.; Visvader, J.E. Cellular mechanisms underlying intertumoral heterogeneity. Trends Cancer 2015, 1, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Chen-Scarabelli, C.; Corsetti, G.; Pasini, E.; Dioguardi, F.S.; Sahni, G.; Narula, J.; Gavazzoni, M.; Patel, H.; Saravolatz, L.; Richard Knight, R.; et al. The spasmogenic effects of the proteasome inhibitor carfilzomib on coronary resistance, vascular tone and reactivity. EBioMedicine 2017, 21, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Flati, V.; Sanità, P.; Pasini, E.; Dioguardi, F.S. Protect and Counter-attack: Nutritional Supplementation with Essential Amino acid Ratios Reduces Doxorubicin–induced Cardiotoxicity in vivo and promote Cancer Cell Death in vitro. J. Cytol. Histol. 2015, 6, 354. [Google Scholar] [CrossRef]
- Bartlett, J.J.; Trivedi, P.C.; Pulinikunnil, T. Autophagic disregulation in doxorubicin cardiomyopathy. Life Sci. 2017, 104, 1–8. [Google Scholar]
- Scognamiglio, R.; Negut, C.; Palisi, M.; Dioguardi, F.S.; Coccato, M.; Iliceto, S. Effects of oral amino acid supplements on cardiac function and remodeling in patients with type 2 diabetes with mild to moderate left ventricular dysfunction. Am. J. Cardiol. 2008, 101, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Henry, C.J. Plasma-free amino acid profiles are predictors of cancer and diabetes development. Nutr. Diabetes 2017, 7, E249. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Cummings, N.E.; Apelo, S.I.A.; Neuman, J.C.; Kasza, I.; Schmidt, B.A.; Cava, E.; Spelta, F.; Tosti, V.; Syed, F.A.; et al. Decreased consumption of branched chain amino acids improves metabolic health. Cell Rep. 2016, 16, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slenz, C.A.; et al. A branched chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, K.; LeBlanc, R.E.; Loh, D.; Schwartz, G.J.; Yu, Y.-H. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes 2007, 56, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Abe, M.; Furukawa, S.; Tokumoto, Y.; Toshimitsu, K.; Ueda, T.; Yamamoto, S.; Hirooka, M.; Kumagi, T.; Hiasa, Y.; et al. Long-term branched-chain amino acid supplementation improves glucose tolerance in patients with nonalcoholic steatohepatitis-related cirrhosis. Intern. Med. 2012, 51, 2151–2155. [Google Scholar] [CrossRef] [PubMed]
- Gojda, J.; Strakovà, R.; Pihalovà, A.; Tùma, P.; Potočkovà, J.; Polàk, J.; Anděl, M. Increased incretin but not insulin response after oral versus intravenous branched chain amino aicds. Ann. Nutr. Metab. 2017, 70, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, H.; Fan, W.; Jin, Q.; Chao, T.; Wu, Y.; Huang, J.; Hao, L.; Yang, X. Leucine supplementation differently modulates branched-chain amino acid catabolism, mitochondrial function and metabolic profiles at the different stage of insulin resistance in rats on high-fat diet. Nutrients 2017, 9, 565. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Hall, M.N. Growth and aging: A common molecular mechanism. Aging 2009, 1, 357–362. [Google Scholar] [CrossRef] [PubMed]
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 2010, 12, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Canfield, J.; Copes, N.; Brito, A.; Rehan, M.; Lipps, D.; Brunquell, J.; Westerheide, S.D.; Bradshaw, P.C. Mechanisms of amino acid-mediated lifespan extension in caenorrhabditis elegans. BMC Genet. 2015, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Giezberg, P.; Daniel, H. Branched-chain amino acids as biomarkers in diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 48–54. [Google Scholar]
- Marchesini, G.; Dioguardi, F.S.; Bianchi, G.P.; Zoli, M.; Bellati, G.; Roffi, L.; Martines, D.; Abbiati, R.; the Italian Multicenter study group. Long-term oral branched-chain amino acid treatment in chronic hepatic encephalopathy. A randomized double-blind, casein-controlled study. J. Hepatol. 1990, 11, 92–101. [Google Scholar] [CrossRef]
- Antoun, S.; Birdsell, L.; Sawyer, M.B.; Venner, P.; Escudier, B.; Baracos, V.E. Association of Skeletal Muscle Wasting with Treatment with Sorafenib in Patients with Advanced Renal Cell Carcinoma: Results From a Placebo-Controlled Study. J. Clin. Oncol. 2010, 28, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Cancer Therapy Evaluation Program: Common Terminology Criteria for Adverse Events v3.0 (CTCAE); US Department of Health and Human Services: Washington, DC, USA, 2006.
- Tremblay, F.; Lavigne, C.; Jacques, H.; Marette, A. Role of Dietary Proteins and Amino Acids in the Pathogenesis of Insulin Resistance. Annu. Rev. Nutr. 2007, 27, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Dioguardi, F.S. Wasting and the substrate-to-energy controlled pathway: A role for insulin resistance and amino acids. Am. J. Cardiol. 2004, 93, 6A–12A. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Pasini, E.; Romano, C.; Calvani, R.; Picca, A.; Marzetti, E.; Flati, V.; Dioguardi, F.S. Body Weight Loss and Tissue Wasting in Late Middle-Aged Mice on Slightly Imbalanced. Essent./Non-essent. Amino Acids Diet. Front. Med. 2018, 17, 136. [Google Scholar] [CrossRef]
- Flati, V.; Corsetti, G.; Pasini, E.; Rufo, A.; Romano, C.; Dioguardi, F.S. Nutrition, Nitrogen Requirements, Exercise and Chemotherapy-Induced Toxicity in Cancer Patients. A puzzle of Contrasting Truths? Anticancer Agents Med. Chem. 2016, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, D.; Hannezo, E.; Hersztberg, S.; Bosveld, F.; Gaugue, I.; Balakireva, M.; Wang, Z.; Cristo, I.; Rigaud, S.U.; Markova, O.; et al. Transmission of cytokines forces via E-cadherin dilution and actomyosin flows. Nature 2017. [Google Scholar] [CrossRef]
- Chen, G.; Wang, J. Threonine metabolism and embryonic stem cell self-renewal. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muto, Y.; Sato, S.; Watanabe, A.; Moriwaki, H.; Suzuki, K.; Kato, A.; Kato, M.; Nakamura, T.; Higuchi, K.; Nishiguchi, S.; et al. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long-term oral supplementation with branched chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol. Res. 2006, 35, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Nishiyama, M.; Ishizaki, S. Branched-Chain Amino Acids Prevent Insulin-Induced Hepatic Tumor Cell Proliferation by Inducing Apoptosis through mTORC1 and mTORC2-Dependent Mechanisms. J. Cell Physiol. 2012, 227, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Gonzàlez, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.C.; Prudner, B.C.; Lange, S.E.S.; Michel, L.S.; Held, J.M.; Van Tine, B.A. Arginine deprivation inhibits the Warburg effect and upregulates glutamine anaplerosis and serine biosynthesis in ASS-1deficient cancers. Cell Rep. 2017, 18, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Rubinzstein, D.C. BCL2L11/BIM. A novel molecular link between autophagy and apoptosis. Autophagy 2013, 9, 104–105. [Google Scholar] [CrossRef] [PubMed]
- Dal Negro, R.W.; Aquilani, R.; Bertacco, S.; Boschi, F.; Micheletto, C.; Tognella, S. Comprehensive effects of supplemented essential amino acids in patients with severe COPD and sarcopenia. Monaldi Arch. Chest Dis. 2010, 73, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Rondanelli, M.; Opizzi, A.; Antoniello, N.; Boschi, F.; Iadarola, P.; Pasini, E.; Aquilani, R.; Dioguardi, F.S. Effect of essential amino acid supplementation on quality of life, amino acid profile and strength in institutionalized elderly patients. Clin. Nutr. 2011, 30, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Pasini, E.; D’Antona, G.; Nisoli, E.; Flati, V.; Assanelli, D.; Dioguardi, F.S.; Bianchi, R. Morphometric changes induced by amino acid supplementation in skeletal and cardiac muscles of old mice. Am. J. Cardiol. 2008, 101, 26E–34E. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, W.J.; Ratamess, N.A.; Volek, J.S.; Hakkinen, K.; Rubin, M.R.; French, D.N.; Gomez, A.L.; McGuigan, M.R.; Scheet, T.P.; Newton, R.U.; et al. The effects of amino acids supplementation on hormonal responses to resistance training overreaching. Metabolism 2006, 55, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, C.; Macciò, A.; Astara, G.; Massa, E.; Dessì, M.; Antoni, G.; Panzone, F.; Serpe, R.; Mantovani, G. Open phase II study on efficacy and safety of an oral amino acid functional cluster supplementation in cancer cachexia. Mediterr. J. Nutr. Metab. 2010, 3, 165–172. [Google Scholar] [CrossRef]
- Scarabelli, T.M.; Pasini, E.; Stephanou, A.; Chen-Scarabelli, C.; Saravolatz, L.; Knight, R.A.; Latchman, D.S.; Gardin, J.M. Nutritional supplementation with mixed essential aminoacids enhance myocyte survival, preserving mitochondrial functional capacity during ischemia-reperfusion injury. Am. J. Cardiol. 2004, 93, 35A–40A. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Vousden, K.H. One carbon, many roads. Cell Death Differ. 2017, 24, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhou, Z.; Deaciuc, I.; Chen, T.; McClain, C.J. Inhibition of Adiponectin Production by Homocysteine: A Potential Mechanism for Alcoholic Liver Disease. Hepatology 2008, 47, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Chim, A.-S.; Fassier, P.; Latino-Martel, P.; Druesne-Pecollo, N.; Zelek, L.; Duverger, L.; Hercberg, S.; Galan, P.; Deschasaux, M.; Touvier, M. Prospective association between alcohol intake and hormone dependent cancer risk: Modulation by dietary fiber intake. Am. J. Clin. Nutr. 2015, 102, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.N.; Giovannucci, E.L. Folic Acid Fortification and Colorectal Cancer Risk. Am. J. Prev. Med. 2014, 46, S65–S72. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J.; Mason, J.B.; Choi, S.-W. Too much folate—A risk factor for cancer and cardiovascular disease? Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Farber, S.; Diamond, L.K. Temporary remission in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; De Flora, S.; Noonan, D.M. Cardiotoxicity of Anticancer Drugs: The Need for Cardio-Oncology and Cardio-Oncological Prevention. J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuvalov, O.; Petukhov, A.; Daks, A.; Fedorova, O.; Vasileva, E.; Barlev, N.A. One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 2017, 8, 23955–23977. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Brit. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ericson, U.C.; Ivarsson, M.I.L.; Sonestedt, E.; Gullberg, B.; Carlson, J.; Olsson, H.; Wirfält, E. Increased breast cancer risk at high plasma folate concentrations among women with the MTHFR 677T allele. Am. J. Clin. Nutr. 2009, 90, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Alcohol Metabolism and Epigenetics Changes. Alcohol. Res. 2013, 35, 6–16. [Google Scholar] [PubMed]
- Gravel, S.-P.; Hulea, L.; Toban, N.; Birman, E.; Blouin, M.-J.; Zakikhani, M.; Zhao, Y.; Topisirovic, I.; St-Pierre, J.; Pollak, M. serine deprivation enhances antineoplastic activity of biguanides. Cancer Res. 2014, 74, 7521–7533. [Google Scholar] [CrossRef] [PubMed]
- Stolzemberg, G. Can an inquiry into the foundation of mathematics tell us anything interesting about mind? In Psychology and Biology of Language and Thought: Essay in Honor of Erich Lenneberg; Academic Press: New York, NY, USA, 1978; pp. 221–269. [Google Scholar]
- Kawaguchi, T.; Shiraishi, K.; Ito, T.; Suzuki, K.; Koreeda, C.; Ohtake, T.; Iwasa, M.; Tokumoto, Y.; Endo, R.; Kawamura, N.H.; et al. Branched-Chain Amino Acids Prevent Hepatocarcinogenesis and Prolong Survival of Patients With Cirrhosis. Clin. Gastroenterol. Hepatol. 2014, 1012–1018.e1. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. MTOR signaling in growth, metabolism and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.F. Time and circumstances: Cancer cell metabolism at various stages of disease progression. Front. Oncol. 2016, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A hallmark of cancer metabolism. Ann. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Still, E.R.; Yuneva, M.O. Hopefully devoted to Q: Targeting glutamine addiction in cancer. Br. J. Cancer 2017, 116, 1375. [Google Scholar] [CrossRef] [PubMed]
- Bezkorovainy, A.; Rafaelson, M.E. (Eds.) Chapter 20. Protein and amino acid metabolism. In Concise Biochemistry; Marcel Dekker Inc.: New York, NY, USA, 1996; pp. 535–571. ISBN 0-8247-9659-4. [Google Scholar]
- Flynn, N.E.; Meininger, C.J.; Haynes, T.E.; Wu, G. The metabolic basis of arginine nutrition and pharmacotherapy. Biomed. Pharmacother. 2002, 56, 427–438. [Google Scholar] [CrossRef]
- Coeffier, M.; Dechelotte, P. The role of glutamine in intensive care unit patients: Mechanisms of action and clinical outcome. Nutr. Rev. 2005, 63, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Aquilani, R.; Zuccarelli, G.C.; Dioguardi, F.S.; Baiardi, P.; Frustaglia, A.; Rutili, C.; Comi, E.; Catani, M.; Iadarola, P.; Viglio, S.; et al. Effects of oral amino acid supplementation on long-term-care-acquired infections in elderly patients. Arch. Gerontol. Geriatr. 2011, 52, e123–e128. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, P.; Dub, T.; Jiang, W.; Peng, L.; Butterworth, R.F. Pathogenesis of hepatic encephalopathy and brain edema in acute liver failure: Role of glutamine redefined. Neurochem. Int. 2012, 60, 690–696. [Google Scholar] [CrossRef] [PubMed]
- James, L.A.; Lunn, P.G.; Middleton, S.; Elia, M. Distribution of glutaminase and glutamine synthase activities in the human gastrointestinal tract. Clin. Sci. 1998, 94, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Deitmer, J.W.; Bröer, A.; Bröer, S. Glutamine efflux from astrocytes is mediated by multiple pathways. J. Neurochem. 2003, 87, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holecek, M. Side effects of long-term glutamine supplementation. J. Parenter. Enter. Nutr. 2013, 37, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Wang, Y.; Watford, M. Glutamine Directly Downregulates Glutamine Synthetase Protein Levels in Mouse C2C12 Skeletal Muscle Myotubes. J. Nutr. 2007, 137, 1357–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Yin, Y.-L.; Li, D.; Kim, S.W.S.; Wu, G. Amino acids and immune function. Brit. J. Nutr. 2007, 98, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phang, J.; Donald, S.P.; Pandhare, J.; Liu, Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 2008, 35, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase) in cancer. Biofactors 2012, 38, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, C.H.; Phang, J.M. Transfer of reducing equivalents into mitochondria by the interconversion of proline and Δ1-pyrroline 5-carboxylate. Arch. Biochem. Biophys. 1983, 225, 25–29. [Google Scholar] [CrossRef]
- Phang, J.M.; Liu, W.; Hancock, C.; Christian, K.J. The proline regulatory axis and cancer. Front. Oncol. 2012, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-L.; Wang, L.-Y.; Yu, Y.-L.; Chen, H.-W.; Sristava, S.; Petrovics, G.; Kung, H.-J. A long noncoding RNA connects c-Myc to tumor metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 18697–18702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. C-MYC mRNA tail tale about glutamine control of transcription. EMBO J. 2017, 36, 1806–1808. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M., Jr. Recent avances in arginine metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Israelsen, W.J.; Dayton, T.L.; Davidson, S.M.; Fiske, B.P.; Hosios, A.M.; Bellinger, G.; Li, J.; Yu, Y.; Sasaki, M.; Horner, J.W.; et al. Vander Heiden MG. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013, 155, 397–409. [Google Scholar] [CrossRef] [PubMed]


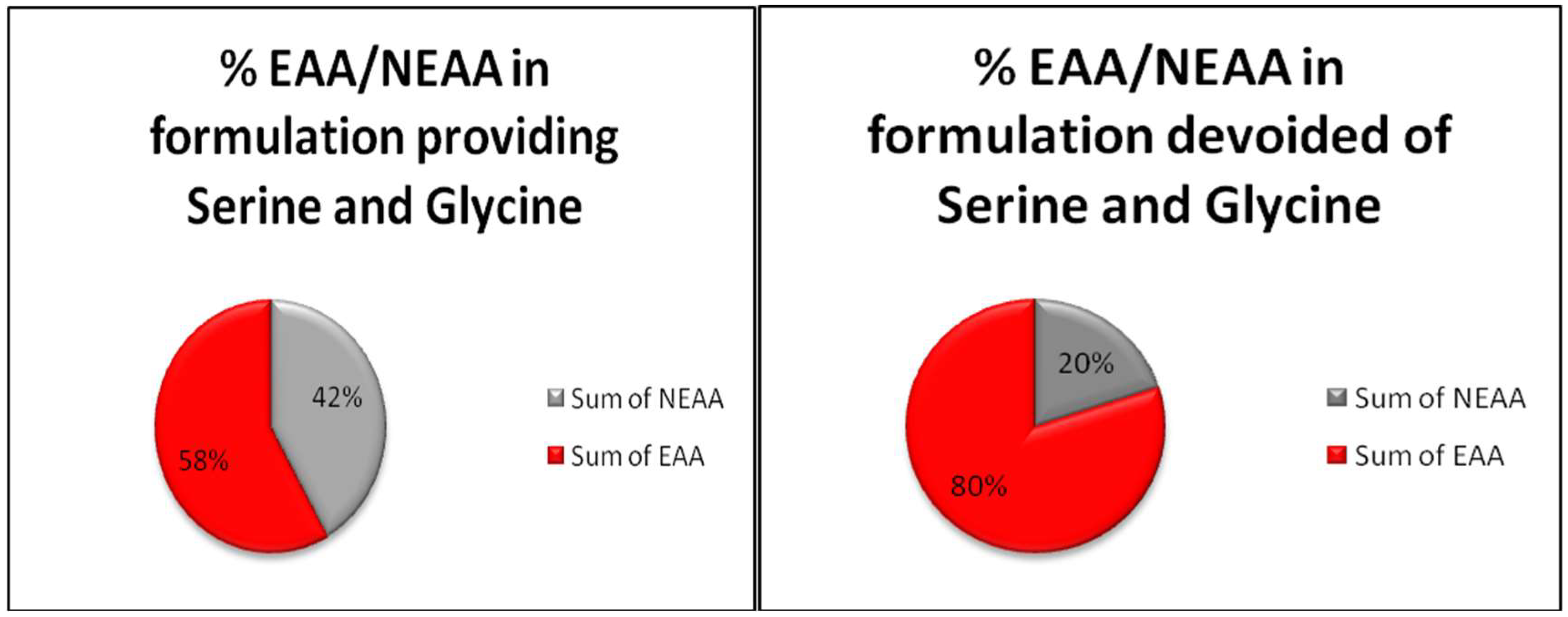{kind=link}
| 100% EAAs (w/w %) | |||||||
| Leucine | Isoleucine | Valine | Histidine | Lysine | Threonine | Methionine * | Phenylalanine |
| 31.25 | 15.625 | 15.625 | 3.75 | 16.25 | 8.75 | 1.25 | 2.5 |
| Tryptophan | Tyrosine ** | Cystine * | |||||
| 0.5 | 0.75 | 3.75 | |||||
| 85% EAAs and 15% NEAAs (w/w %) | |||||||
| Leucine | Isoleucine | Valine | Histidine | Lysine | Threonine | Methionine * | Phenylalanine |
| 13.53 | 9.65 | 9.65 | 11.60 | 11.60 | 8.70 | 4.35 | 7.73 |
| Tryptophan | Tyrosine ** | Cystine * | Serine | N-Acetyl Cysteine * | Ornithine-α Ketoglutarate | ||
| 3.38 | 5.80 | 8.20 | 2.42 | 0.97 | 2.42 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dioguardi, F.S.; Flati, V.; Corsetti, G.; Pasini, E.; Romano, C. Is the Response of Tumours Dependent on the Dietary Input of Some Amino Acids or Ratios among Essential and Non-Essential Amino Acids? All That Glitters Is Not Gold. Int. J. Mol. Sci. 2018, 19, 3631. https://doi.org/10.3390/ijms19113631
Dioguardi FS, Flati V, Corsetti G, Pasini E, Romano C. Is the Response of Tumours Dependent on the Dietary Input of Some Amino Acids or Ratios among Essential and Non-Essential Amino Acids? All That Glitters Is Not Gold. International Journal of Molecular Sciences. 2018; 19(11):3631. https://doi.org/10.3390/ijms19113631
Chicago/Turabian StyleDioguardi, Francesco S., Vincenzo Flati, Giovanni Corsetti, Evasio Pasini, and Claudia Romano. 2018. "Is the Response of Tumours Dependent on the Dietary Input of Some Amino Acids or Ratios among Essential and Non-Essential Amino Acids? All That Glitters Is Not Gold" International Journal of Molecular Sciences 19, no. 11: 3631. https://doi.org/10.3390/ijms19113631
APA StyleDioguardi, F. S., Flati, V., Corsetti, G., Pasini, E., & Romano, C. (2018). Is the Response of Tumours Dependent on the Dietary Input of Some Amino Acids or Ratios among Essential and Non-Essential Amino Acids? All That Glitters Is Not Gold. International Journal of Molecular Sciences, 19(11), 3631. https://doi.org/10.3390/ijms19113631