Programming of Vascular Dysfunction in the Intrauterine Milieu of Diabetic Pregnancies

 , and
, and
Abstract
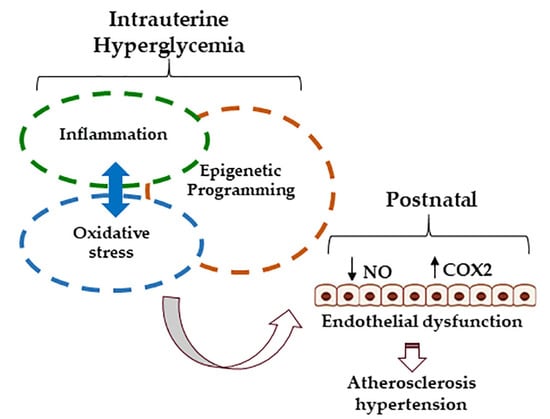
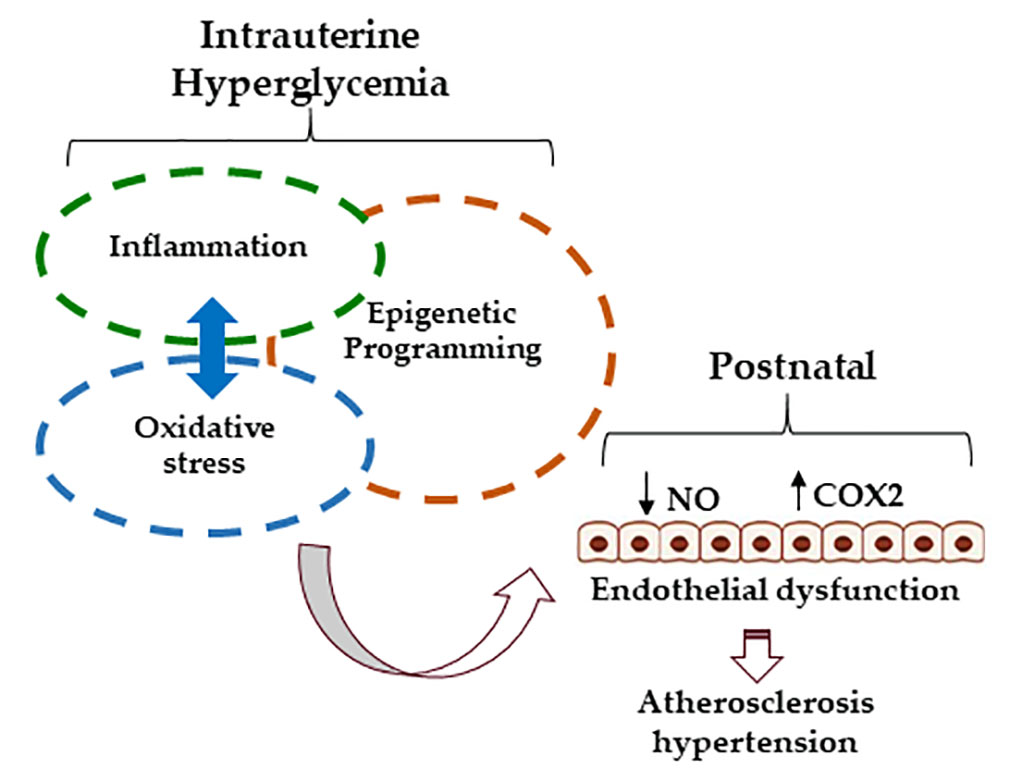
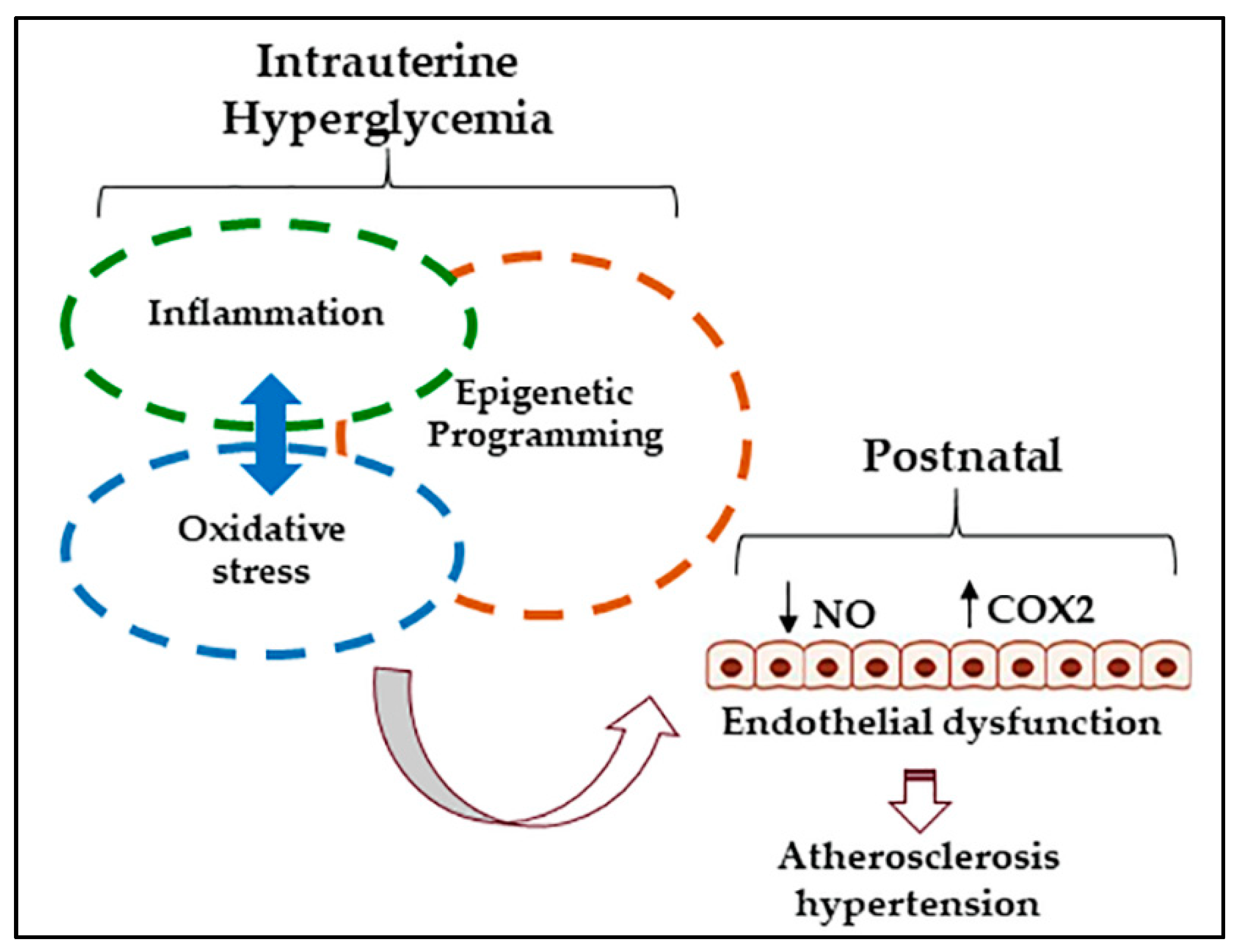
:
1. Prevalence and Pathophysiology
2. Intrauterine Metabolic Milieu
3. Cardiovascular Disease (CVD) Risk Factors in the Offspring
4. Vascular Dysfunction: Human studies
5. Vascular Dysfunction: Animal Studies
6. Mechanisms: Oxidative Stress
7. Mechanisms: Inflammation
8. Mechanisms: Epigenetics
9. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farrar, D. Hyperglycemia in pregnancy: prevalence, impact, and management challenges. Int. J. Women’s Health 2016, 8, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Weinert, L.S. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy: comment to the international association of diabetes and pregnancy study groups consensus panel. Diabetes Care 2010, 33, e97. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.A.; Hadden, D.R.; Maresh, M.; Deerochanawong, C.; Dyer, A.R.; Metzger, B.E.; Lowe, L.P.; Coustan, D.R.; Hod, M.; Oats, J.J.; et al. Frequency of gestational diabetes mellitus at collaborating centers based on IADPSG consensus panel-recommended criteria: the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. Diabetes Care 2012, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Oteng-Ntim, E.; Mononen, S.; Sawicki, O.; Seed, P.T.; Bick, D.; Poston, L. Interpregnancy weight change and adverse pregnancy outcomes: a systematic review and meta-analysis. BMJ Open 2018, 8, e018778. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.S.; Shields, M.; Laviolette, M.; Craig, C.L.; Janssen, I.; Connor Gorber, S. Fitness of Canadian children and youth: results from the 2007-2009 Canadian Health Measures Survey. Health Rep. 2010, 21, 7–20. [Google Scholar] [PubMed]
- Hales, C.M.; Fryar, C.D.; Carroll, M.D.; Freedman, D.S.; Aoki, Y.; Ogden, C.L. Differences in obesity prevalence by demographic characteristics and urbanization level among adults in the united states, 2013-2016. JAMA 2018, 319, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.; Mulla, Z.D.; Plavsic, S.K. Outcomes of women delivering at very advanced maternal age. J. Women’s Health 2018, 27, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Damm, P.; Houshmand-Oeregaard, A.; Kelstrup, L.; Lauenborg, J.; Mathiesen, E.R.; Clausen, T.D. Gestational diabetes mellitus and long-term consequences for mother and offspring: a view from Denmark. Diabetologia 2016, 59, 1396–1399. [Google Scholar] [CrossRef] [PubMed]
- Kautzky-Willer, A.; Prager, R.; Waldhausl, W.; Pacini, G.; Thomaseth, K.; Wagner, O.F.; Ulm, M.; Streli, C.; Ludvik, B. Pronounced insulin resistance and inadequate beta-cell secretion characterize lean gestational diabetes during and after pregnancy. Diabetes Care 1997, 20, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Snider, F.; Cross, J.C. Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy. Endocrinology 2009, 150, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.; Huang, C. Participation of Akt, menin, and p21 in pregnancy-induced beta-cell proliferation. Endocrinology 2011, 152, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Baecker, A.; Song, Y.; Kiely, M.; Liu, S.; Zhang, C. Adipokine levels during the first or early second trimester of pregnancy and subsequent risk of gestational diabetes mellitus: A systematic review. Metab. Clin. Exp. 2015, 64, 756–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirwan, J.P.; Hauguel-de Mouzon, S.; Lepercq, J.; Challier, J.C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes 2002, 51, 2207–2213. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Dostalova, I.; Bartlova, M.; Lacinova, Z.; Ticha, I.; Krejci, V.; Springer, D.; Kleibl, Z.; Haluzik, M. Expression of adipokines and estrogen receptors in adipose tissue and placenta of patients with gestational diabetes mellitus. Mol. Cell. Endocrinol. 2010, 314, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, G.; Cardenas, I.; Abrahams, V.; Guller, S. Inflammation and pregnancy: the role of the immune system at the implantation site. Ann. N. Y. Acad. Sci. 2011, 1221, 80–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotechini, T.; Graham, C.H. Aberrant maternal inflammation as a cause of pregnancy complications: A potential therapeutic target? Placenta 2015, 36, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Porto, N.P.; Juca, D.M.; Lahlou, S.; Coelho-de-Souza, A.N.; Duarte, G.P.; Magalhaes, P.J. Effects of K+channels inhibitors on the cholinergic relaxation of the isolated aorta of adult offspring rats exposed to maternal diabetes. Exp. Clin. Endocrinol. Diabetes 2010, 118, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.B.; Ferrara, A.; Ehrlich, S.F.; Brown, S.D.; Hedderson, M.M. Risk of large-for-gestational-age newborns in women with gestational diabetes by race and ethnicity and body mass index categories. Obstet. Gynecol. 2013, 121, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Bodmer-Roy, S.; Morin, L.; Cousineau, J.; Rey, E. Pregnancy outcomes in women with and without gestational diabetes mellitus according to the International Association of the Diabetes and Pregnancy Study Groups criteria. Obstet. Gynecol. 2012, 120, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Negrato, C.A.; Mattar, R.; Gomes, M.B. Adverse pregnancy outcomes in women with diabetes. Diabetol. Metab. Syndr. 2012, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer-Graf, U.M.; Meitzner, K.; Ortega-Senovilla, H.; Graf, K.; Vetter, K.; Abou-Dakn, M.; Herrera, E. Differences in the implications of maternal lipids on fetal metabolism and growth between gestational diabetes mellitus and control pregnancies. Diabet. Med. 2011, 28, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Di Cianni, G.; Miccoli, R.; Volpe, L.; Lencioni, C.; Ghio, A.; Giovannitti, M.G.; Cuccuru, I.; Pellegrini, G.; Chatzianagnostou, K.; Boldrini, A.; et al. Maternal triglyceride levels and newborn weight in pregnant women with normal glucose tolerance. Diabet. Med. 2005, 22, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Fang, Z.; Yang, D.; Liu, Q. Correlation between the inflammatory factors and adipocytokines with gestational diabetes mellitus and their change in puerperium. Zhonghua Fu Chan Ke Za Zhi 2012, 47, 436–439. [Google Scholar] [PubMed]
- Perng, W.; Rifas-Shiman, S.L.; McCulloch, S.; Chatzi, L.; Mantzoros, C.; Hivert, M.F.; Oken, E. Associations of cord blood metabolites with perinatal characteristics, newborn anthropometry, and cord blood hormones in project viva. Metab. Clin. Exp. 2017, 76, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; McIntyre, H.D.; Cruickshank, J.K.; McCance, D.R.; Dyer, A.R.; Metzger, B.E.; Lowe, L.P.; Trimble, E.R.; Coustan, D.R.; Hadden, D.R.; et al. The hyperglycemia and adverse pregnancy outcome study: associations of GDM and obesity with pregnancy outcomes. Diabetes Care 2012, 35, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Manderson, J.G.; Mullan, B.; Patterson, C.C.; Hadden, D.R.; Traub, A.I.; McCance, D.R. Cardiovascular and metabolic abnormalities in the offspring of diabetic pregnancy. Diabetologia 2002, 45, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Bunt, J.C.; Tataranni, P.A.; Salbe, A.D. Intrauterine exposure to diabetes is a determinant of hemoglobin A(1)c and systolic blood pressure in pima Indian children. J. Clin. Endocrinol. Metab. 2005, 90, 3225–3229. [Google Scholar] [CrossRef] [PubMed]
- Landon, M.B.; Rice, M.M.; Varner, M.W.; Casey, B.M.; Reddy, U.M.; Wapner, R.J.; Rouse, D.J.; Biggio, J.R.J.; Thorp, J.M.; Chien, E.K.; et al. Mild gestational diabetes mellitus and long-term child health. Diabetes Care 2015, 38, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.H.; Ma, R.C.W.; Ozaki, R.; Li, A.M.; Chan, M.H.M.; Yuen, L.Y.; Lao, T.T.H.; Yang, X.; Ho, C.S.; Tutino, G.E.; et al. In utero exposure to maternal hyperglycemia increases childhood cardiometabolic risk in offspring. Diabetes Care 2017, 40, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Hanson, R.L.; Lindsay, R.S.; Pettitt, D.J.; Imperatore, G.; Gabir, M.M.; Roumain, J.; Bennett, P.H.; Knowler, W.C. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes 2000, 49, 2208–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, W.H.; Ma, R.C.; Yip, G.W.; Yang, X.; Li, A.M.; Ko, G.T.; Lao, T.T.; Chan, J.C. The association between in utero hyperinsulinemia and adolescent arterial stiffness. Diabetes Res. Clin. Pract. 2012, 95, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Akcakus, M.; Koklu, E.; Baykan, A.; Yikilmaz, A.; Coskun, A.; Gunes, T.; Kurtoglu, S.; Narin, N. Macrosomic newborns of diabetic mothers are associated with increased aortic intima-media thickness and lipid concentrations. Horm. Res. 2007, 67, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Larion, S.; Mintz, J.D.; Belin de Chantemele, E.J.; Fulton, D.J.; Stepp, D.W. Genetic deletion of NADPH oxidase 1 rescues microvascular function in mice with metabolic disease. Circ. Res. 2017, 121, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Haas, D.M.; Saha, C.K.; Dimeglio, L.A.; Ingram, D.A.; Haneline, L.S. Gestational diabetes mellitus alters maternal and neonatal circulating endothelial progenitor cell subsets. Am. J. Obstet. Gynecol. 2011, 204, 254.e8–254.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.A.; Crume, T.L.; Maligie, M.A.; Dabelea, D. Cardiovascular risk factors in children exposed to maternal diabetes in utero. Diabetologia 2011, 54, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Engerman, R.L.; Kern, T.S. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 1987, 36, 808–812. [Google Scholar] [CrossRef] [PubMed]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tomo, P.; Lanuti, P.; di Pietro, N.; Baldassarre, M.P.A.; Marchisio, M.; Pandolfi, A.; Consoli, A.; Formoso, G. Liraglutide mitigates TNF-alpha induced pro-atherogenic changes and microvesicle release in HUVEC from diabetic women. Diabetes Metab. Res. Rev. 2017, 33. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Hiden, U.; Desoye, G.; Froehlich, J.; Hauguel-de Mouzon, S.; Jawerbaum, A. The role of oxidative stress in the pathophysiology of gestational diabetes mellitus. Antioxid. Redox Signal. 2011, 15, 3061–3100. [Google Scholar] [CrossRef] [PubMed]
- Aljunaidy, M.M.; Morton, J.S.; Kirschenman, R.; Phillips, T.; Case, C.P.; Cooke, C.M.; Davidge, S.T. Maternal treatment with a placental-targeted antioxidant (MitoQ) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmacol. Res. 2018, 134, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A.; Camm, E.J.; Niu, Y.; Richter, H.G.; Blanco, C.E.; Gottschalk, R.; Blake, E.Z.; Horder, K.A.; Thakor, A.S.; Hansell, J.A.; et al. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS ONE 2012, 7, e31017. [Google Scholar] [CrossRef] [PubMed]
- Kinalski, M.; Sledziewski, A.; Telejko, B.; Kowalska, I.; Kretowski, A.; Zarzycki, W.; Kinalska, I. Lipid peroxidation, antioxidant defence and acid-base status in cord blood at birth: the influence of diabetes. Horm. Metab. Res. 2001, 33, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Vervaart, P.P.; Permezel, M.; Georgiou, H.M.; Rice, G.E. Altered placental oxidative stress status in gestational diabetes mellitus. Placenta 2004, 25, 78–84. [Google Scholar] [CrossRef]
- Lappas, M.; Mitton, A.; Permezel, M. In response to oxidative stress, the expression of inflammatory cytokines and antioxidant enzymes are impaired in placenta, but not adipose tissue, of women with gestational diabetes. J. Endocrinol. 2010, 204, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Chapple, S.J.; Patel, B.; Puszyk, W.; Sugden, D.; Yin, X.; Mayr, M.; Siow, R.C.; Mann, G.E. Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. Diabetes 2013, 62, 4088–4097. [Google Scholar] [CrossRef] [PubMed]
- Madazli, R.; Tuten, A.; Calay, Z.; Uzun, H.; Uludag, S.; Ocak, V. The incidence of placental abnormalities, maternal and cord plasma malondialdehyde and vascular endothelial growth factor levels in women with gestational diabetes mellitus and nondiabetic controls. Gynecol. Obstet. Invest. 2008, 65, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Biri, A.; Onan, A.; Devrim, E.; Babacan, F.; Kavutcu, M.; Durak, I. Oxidant status in maternal and cord plasma and placental tissue in gestational diabetes. Placenta 2006, 27, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Castrejon, M.; Powell, T.L. Placental Nutrient Transport in Gestational Diabetic Pregnancies. Front. Endocrinol. 2017, 8, 306. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, T.; Klepcyk, P.; Liu, S.; Panko, L.; Liu, S.; Gibbs, E.M.; Friedman, J.E. Effects of overexpression of human GLUT4 gene on maternal diabetes and fetal growth in spontaneous gestational diabetic C57BLKS/J Lepr(db/+) mice. Diabetes 1999, 48, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.A.; Ramirez-Perez, F.I.; Pollock, K.E.; Talton, O.O.; Foote, C.A.; Reyes-Aldasoro, C.C.; Wu, H.H.; Ji, T.; Martinez-Lemus, L.A.; Schulz, L.C. Maternal Hyperleptinemia Is Associated with Male Offspring’s Altered Vascular Function and Structure in Mice. PLoS ONE 2016, 11, e0155377. [Google Scholar] [CrossRef] [PubMed]
- Magee, T.R.; Ross, M.G.; Wedekind, L.; Desai, M.; Kjos, S.; Belkacemi, L. Gestational diabetes mellitus alters apoptotic and inflammatory gene expression of trophobasts from human term placenta. J. Diabetes Complications 2014, 28, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aye, I.L.; Jansson, T.; Powell, T.L. Interleukin-1beta inhibits insulin signaling and prevents insulin-stimulated system A amino acid transport in primary human trophoblasts. Mol. Cell. Endocrinol. 2013, 381, 46–55. [Google Scholar] [CrossRef] [PubMed]
- De Sa, F.G.; de Queiroz, D.B.; Ramos-Alves, F.E.; Santos-Rocha, J.; da Silva, O.A.; Moreira, H.S.; Leal, G.A.; da Rocha, M.A.; Duarte, G.P.; Xavier, F.E. Hyperglycaemia in pregnant rats causes sex-related vascular dysfunction in adult offspring: role of cyclooxygenase-2. Exp. Physiol. 2017, 102, 1019–1036. [Google Scholar] [CrossRef] [PubMed]
- Vessieres, E.; Dib, A.; Bourreau, J.; Lelievre, E.; Custaud, M.A.; Lelievre-Pegorier, M.; Loufrani, L.; Henrion, D.; Fassot, C. Long lasting microvascular tone alteration in rat offspring exposed in utero to maternal hyperglycaemia. PLoS ONE 2016, 11, e0146830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segar, E.M.; Norris, A.W.; Yao, J.R.; Hu, S.; Koppenhafer, S.L.; Roghair, R.D.; Segar, J.L.; Scholz, T.D. Programming of growth, insulin resistance and vascular dysfunction in offspring of late gestation diabetic rats. Clin. Sci. 2009, 117, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holemans, K.; Gerber, R.T.; Meurrens, K.; De Clerck, F.; Poston, L.; Van Assche, F.A. Streptozotocin diabetes in the pregnant rat induces cardiovascular dysfunction in adult offspring. Diabetologia 1999, 42, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wichi, R.B.; Souza, S.B.; Casarini, D.E.; Morris, M.; Barreto-Chaves, M.L.; Irigoyen, M.C. Increased blood pressure in the offspring of diabetic mothers, American journal of physiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1129–R1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Queiroz, D.B.; Sastre, E.; Caracuel, L.; Callejo, M.; Xavier, F.E.; Blanco-Rivero, J.; Balfagon, G. Alterations in perivascular innervation function in mesenteric arteries from offspring of diabetic rats. Br. J. Pharmacol. 2015, 172, 4699–4713. [Google Scholar] [CrossRef] [PubMed]
- Katkhuda, R.; Peterson, E.S.; Roghair, R.D.; Norris, A.W.; Scholz, T.D.; Segar, J.L. Sex-specific programming of hypertension in offspring of late-gestation diabetic rats. Pediatr. Res. 2012, 72, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Alves, F.E.; de Queiroz, D.B.; Santos-Rocha, J.; Duarte, G.P.; Xavier, F.E. Effect of age and COX-2-derived prostanoids on the progression of adult vascular dysfunction in the offspring of diabetic rats. Br. J. Pharmacol. 2012, 166, 2198–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadif, R.; Dilworth, M.R.; Sibley, C.P.; Baker, P.N.; Davidge, S.T.; Gibson, J.M.; Aplin, J.D.; Westwood, M. The maternal environment programs postnatal weight gain and glucose tolerance of male offspring, but placental and fetal growth are determined by fetal genotype in the Lepr(db/+) model of gestational diabetes. Endocrinology 2015, 156, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Plows, J.F.; Yu, X.; Broadhurst, R.; Vickers, M.H.; Tong, C.; Zhang, H.; Qi, H.; Stanley, J.L.; Baker, P.N. Absence of a gestational diabetes phenotype in the LepR(db/+) mouse is independent of control strain, diet, misty allele, or parity. Sci. Rep. 2017, 7, 45130. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Zheng, Q.; Ford, S.P.; Nathanielsz, P.W.; Ren, J. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J. Mol. Cell. Cardiol. 2013, 55, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Sallam, N.; Laher, I. Exercise Modulates Oxidative Stress and Inflammation in Aging and Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 7239639. [Google Scholar] [CrossRef] [PubMed]
- Amrithraj, A.I.; Kodali, A.; Nguyen, L.; Teo, A.K.K.; Chang, C.W.; Karnani, N.; Ng, K.L.; Gluckman, P.D.; Chong, Y.S.; Stunkel, W. Gestational Diabetes Alters Functions in Offspring’s Umbilical Cord Cells With Implications for Cardiovascular Health. Endocrinology 2017, 158, 2102–2112. [Google Scholar] [CrossRef] [PubMed]
- Floris, I.; Descamps, B.; Vardeu, A.; Mitic, T.; Posadino, A.M.; Shantikumar, S.; Sala-Newby, G.; Capobianco, G.; Mangialardi, G.; Howard, L.; et al. Gestational diabetes mellitus impairs fetal endothelial cell functions through a mechanism involving microRNA-101 and histone methyltransferase enhancer of zester homolog-2. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Rohrbach, A.; Borns, K.; Hillemanns, P.; Feng, L.; Hubel, C.A.; von Versen-Hoynck, F. Vitamin D rescues dysfunction of fetal endothelial colony forming cells from individuals with gestational diabetes. Placenta 2015, 36, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Ingram, D.A.; Lien, I.Z.; Mead, L.E.; Estes, M.; Prater, D.N.; Derr-Yellin, E.; DiMeglio, L.A.; Haneline, L.S. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes 2008, 57, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Sultan, S.A.; Liu, W.; Peng, Y.; Roberts, W.; Whitelaw, D.; Graham, A.M. The Role of Maternal Gestational Diabetes in Inducing Fetal Endothelial Dysfunction. J. Cell. Physiol. 2015, 230, 2695–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fulvio, P.; Pandolfi, A.; Formoso, G.; Di Silvestre, S.; Di Tomo, P.; Giardinelli, A.; De Marco, A.; Di Pietro, N.; Taraborrelli, M.; Sancilio, S.; et al. Features of endothelial dysfunction in umbilical cord vessels of women with gestational diabetes. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Saez, T.; Salsoso, R.; Leiva, A.; Toledo, F.; de Vos, P.; Faas, M.; Sobrevia, L. Human umbilical vein endothelium-derived exosomes play a role in foetoplacental endothelial dysfunction in gestational diabetes mellitus. Biochim. Biophys. Acta 2018, 1864, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Chen, J.; Das, K.C.; Kavdia, M. Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc. Diabetol. 2013, 12, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ategbo, J.M.; Grissa, O.; Yessoufou, A.; Hichami, A.; Dramane, K.L.; Moutairou, K.; Miled, A.; Grissa, A.; Jerbi, M.; Tabka, Z.; et al. Modulation of adipokines and cytokines in gestational diabetes and macrosomia. J. Clin. Endocrinol. Metab. 2006, 91, 4137–4143. [Google Scholar] [CrossRef] [PubMed]
- Kuzmicki, M.; Telejko, B.; Zonenberg, A.; Szamatowicz, J.; Kretowski, A.; Nikolajuk, A.; Laudanski, P.; Gorska, M. Circulating pro- and anti-inflammatory cytokines in Polish women with gestational diabetes. Horm. Metab. Res. 2008, 40, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Lobo, T.F.; Borges, C.M.; Mattar, R.; Gomes, C.P.; de Angelo, A.G.S.; Pendeloski, K.P.T.; Daher, S. Impaired Treg and NK cells profile in overweight women with gestational diabetes mellitus. Am. J. Reprod. Immunol. 2018, 79. [Google Scholar] [CrossRef] [PubMed]
- Radaelli, T.; Varastehpour, A.; Catalano, P.; Hauguel-de Mouzon, S. Gestational diabetes induces placental genes for chronic stress and inflammatory pathways. Diabetes 2003, 52, 2951–2958. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.L.; Lager, S.; Ramirez, V.I.; Gaccioli, F.; Dudley, D.J.; Jansson, T.; Powell, T.L. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol. Reprod. 2014, 90, 129. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.L.; Jansson, T.; Powell, T.L. TNF-alpha stimulates System A amino acid transport in primary human trophoblast cells mediated by p38 MAPK signaling. Physiol. Rep. 2015, 3. [Google Scholar]
- Piconi, L.; Quagliaro, L.; Da Ros, R.; Assaloni, R.; Giugliano, D.; Esposito, K.; Szabo, C.; Ceriello, A. Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: the role of poly(ADP-ribose) polymerase. J. Thromb. Haemost. 2004, 2, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costantino, S.; Ambrosini, S.; Paneni, F. The epigenetic landscape in the cardiovascular complications of diabetes. J. Endocrinol. Invest. 2018. [CrossRef] [PubMed]
- Houde, A.A.; Guay, S.P.; Desgagne, V.; Hivert, M.F.; Baillargeon, J.P.; St-Pierre, J.; Perron, P.; Gaudet, D.; Brisson, D.; Bouchard, L. Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status. Epigenetics 2013, 8, 1289–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petropoulos, S.; Guillemin, C.; Ergaz, Z.; Dimov, S.; Suderman, M.; Weinstein-Fudim, L.; Ornoy, A.; Szyf, M. Gestational Diabetes Alters Offspring DNA Methylation Profiles in Human and Rat: Identification of Key Pathways Involved in Endocrine System Disorders, Insulin Signaling, Diabetes Signaling, and ILK Signaling. Endocrinology 2015, 156, 2222–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blue, E.K.; Sheehan, B.M.; Nuss, Z.V.; Boyle, F.A.; Hocutt, C.M.; Gohn, C.R.; Varberg, K.M.; McClintick, J.N.; Haneline, L.S. Epigenetic Regulation of Placenta-Specific 8 Contributes to Altered Function of Endothelial Colony-Forming Cells Exposed to Intrauterine Gestational Diabetes Mellitus. Diabetes 2015, 64, 2664–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Piaggi, P.; Traurig, M.; Bogardus, C.; Knowler, W.C.; Baier, L.J.; Hanson, R.L. Differential methylation of genes in individuals exposed to maternal diabetes in utero. Diabetologia 2017, 60, 645–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.A.; Kechris, K.; Dabelea, D. Exposure to Maternal Diabetes in Utero and DNA Methylation Patterns in the Offspring. Immunometabolism 2013, 1, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G. MicroRNAs and Endothelial (Dys) Function. J. Cell. Physiol. 2016, 231, 1638–1644. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Hoelscher, M.; Cattelan, A.; Schmitz, T.; Proebsting, S.; Wenzel, D.; Vosen, S.; Franklin, B.S.; Fleischmann, B.K.; et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation 2013, 128, 2026–2038. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.X.; Zeng, D.Y.; Li, R.T.; Pang, R.P.; Yang, H.; Hu, Y.L.; Zhang, Q.; Jiang, Y.; Huang, L.Y.; Tang, Y.B.; et al. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension 2012, 60, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, X.; Yang, J.; Lin, Y.; Xu, D.Z.; Lu, Q.; Deitch, E.A.; Huo, Y.; Delphin, E.S.; Zhang, C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ. Res. 2009, 105, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, J.; Nema, V.; Dhas, Y.; Mishra, N. Role of MicroRNAs in Type 2 Diabetes and Associated Vascular Complications. Biochimie 2017, 139, 9–19. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Age Group | Examined Tissue/ParameTer | Phenotype | Reference |
|---|---|---|---|
| At birth | HUVEC | Reduced proliferation, tubule formation and migration capacity, increased apoptosis, higher superoxide production and markers of oxidative stress, decreased NO availability, increased monocyte adhesion, heightened inflammatory response | [38,39,40,41,42,43,44,45] |
| ECFC | Reduced ECFC number and impaired proliferation, migration and tubule formation | [46,47,48] | |
| Cord blood | Increased markers of lipid peroxidation, change in DNA methylation status of genes involved in lipid metabolism and atherosclerosis | [49,50,51] | |
| Infants (3–5 days old) | Abdominal aorta | Greater aortic intima-media thickness after adjustment for body weight | [30] |
| Children/adolescent (2–18 years old) | Blood | Increased triglycerides, increased cell adhesion molecules (CAM), change in DNA methylation pattern of genes involved in atherosclerosis and CAM expression | [25,26,35,52] |
| Haemodynamics | High systolic blood pressure, positive association between umbilical insulin levels and augmentation index | [27,29,35] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sallam, N.A.; Palmgren, V.A.C.; Singh, R.D.; John, C.M.; Thompson, J.A. Programming of Vascular Dysfunction in the Intrauterine Milieu of Diabetic Pregnancies. Int. J. Mol. Sci. 2018, 19, 3665. https://doi.org/10.3390/ijms19113665
Sallam NA, Palmgren VAC, Singh RD, John CM, Thompson JA. Programming of Vascular Dysfunction in the Intrauterine Milieu of Diabetic Pregnancies. International Journal of Molecular Sciences. 2018; 19(11):3665. https://doi.org/10.3390/ijms19113665
Chicago/Turabian StyleSallam, Nada A., Victoria A. C. Palmgren, Radha D. Singh, Cini M. John, and Jennifer A. Thompson. 2018. "Programming of Vascular Dysfunction in the Intrauterine Milieu of Diabetic Pregnancies" International Journal of Molecular Sciences 19, no. 11: 3665. https://doi.org/10.3390/ijms19113665
APA StyleSallam, N. A., Palmgren, V. A. C., Singh, R. D., John, C. M., & Thompson, J. A. (2018). Programming of Vascular Dysfunction in the Intrauterine Milieu of Diabetic Pregnancies. International Journal of Molecular Sciences, 19(11), 3665. https://doi.org/10.3390/ijms19113665