Methyl-β-Cyclodextrin Impairs the Phosphorylation of the β2 Subunit of L-Type Calcium Channels and Cytosolic Calcium Homeostasis in Mature Cerebellar Granule Neurons



Abstract
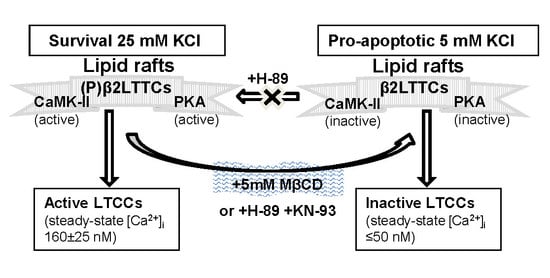
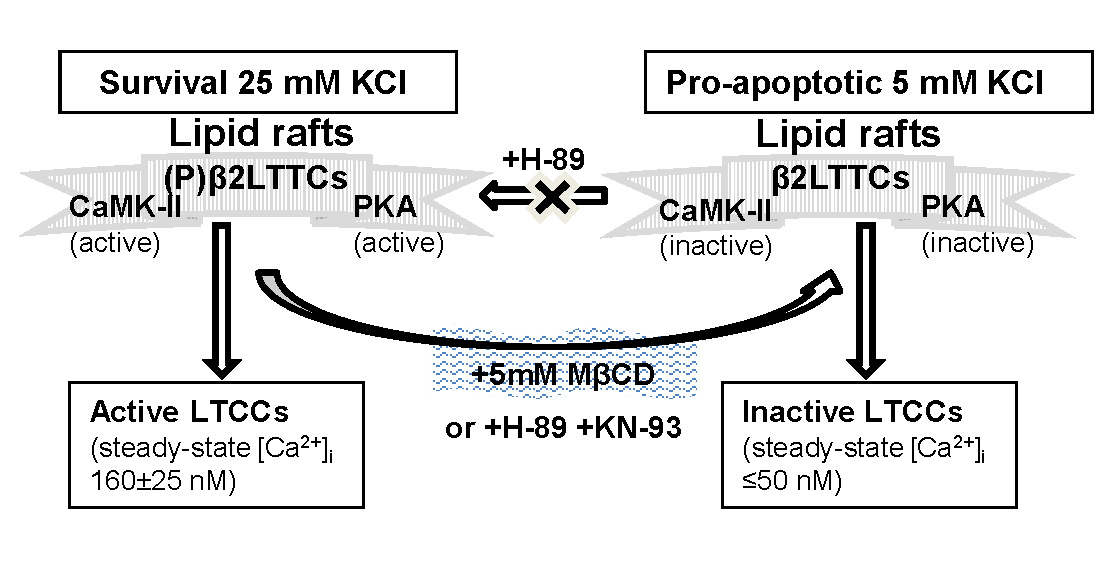
:
1. Introduction
2. Results
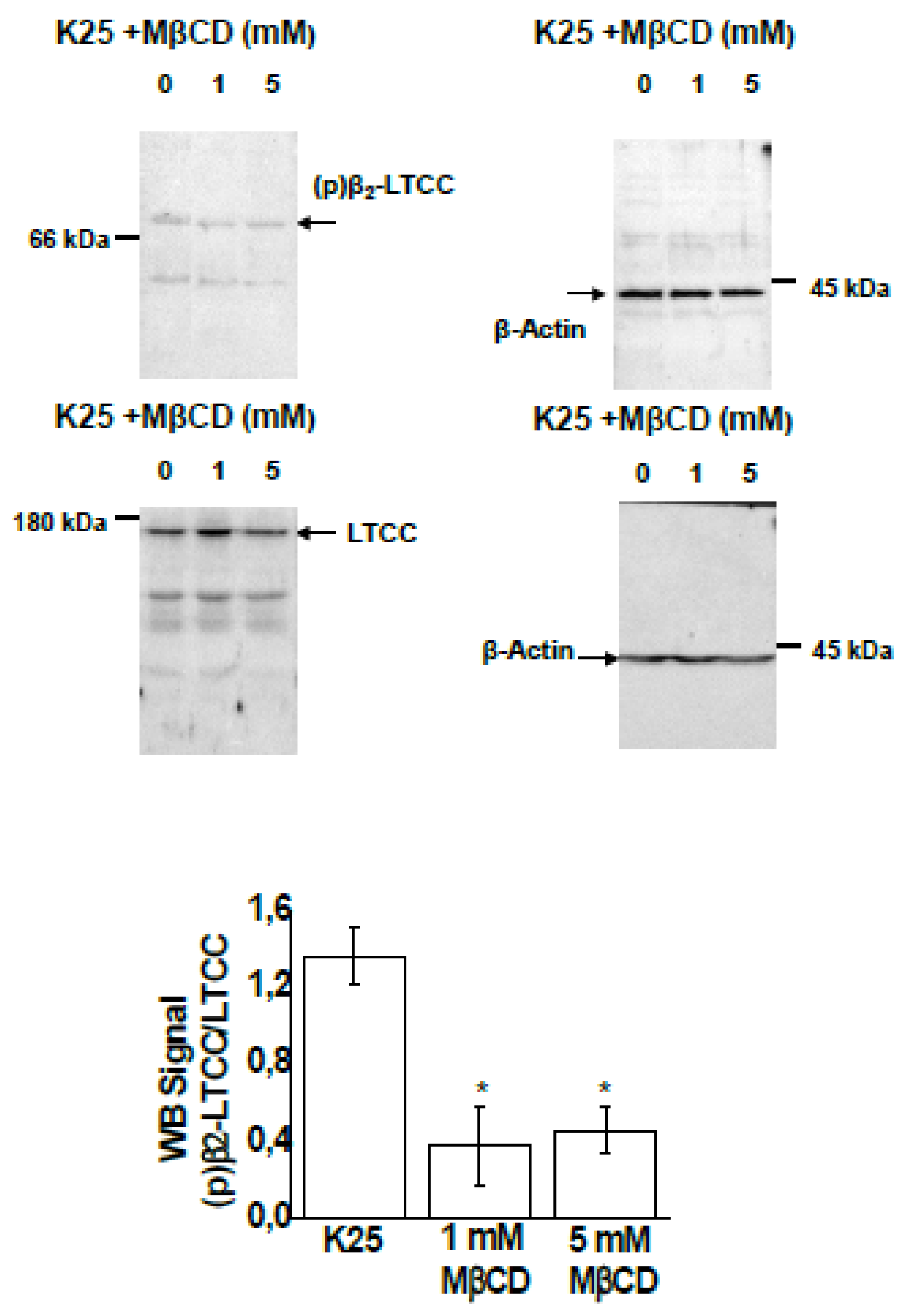
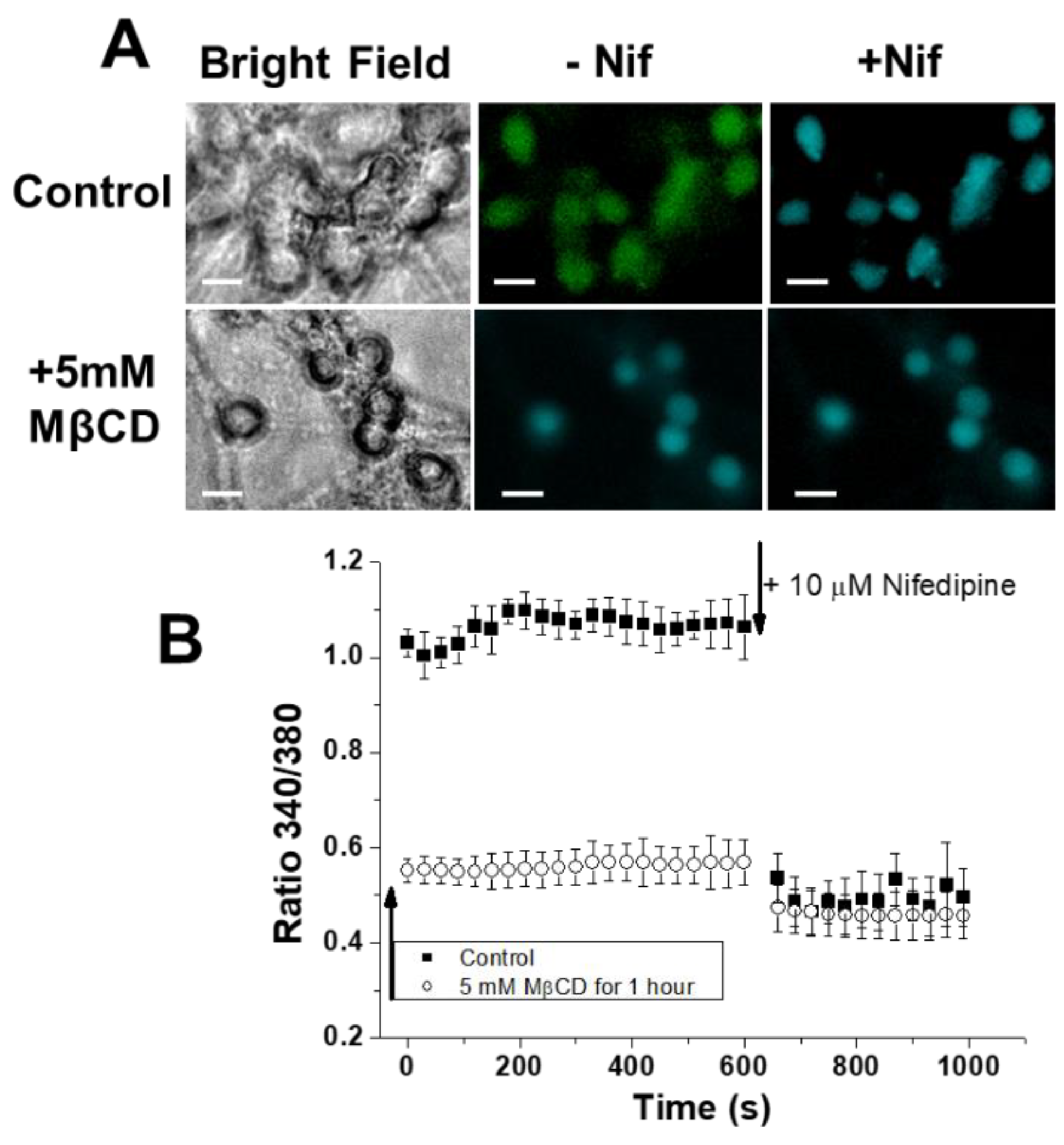
2.1. Treatment of CGNs with MβCD Decreases the Phosphorylation Level of the β2 Subunit of LTCC
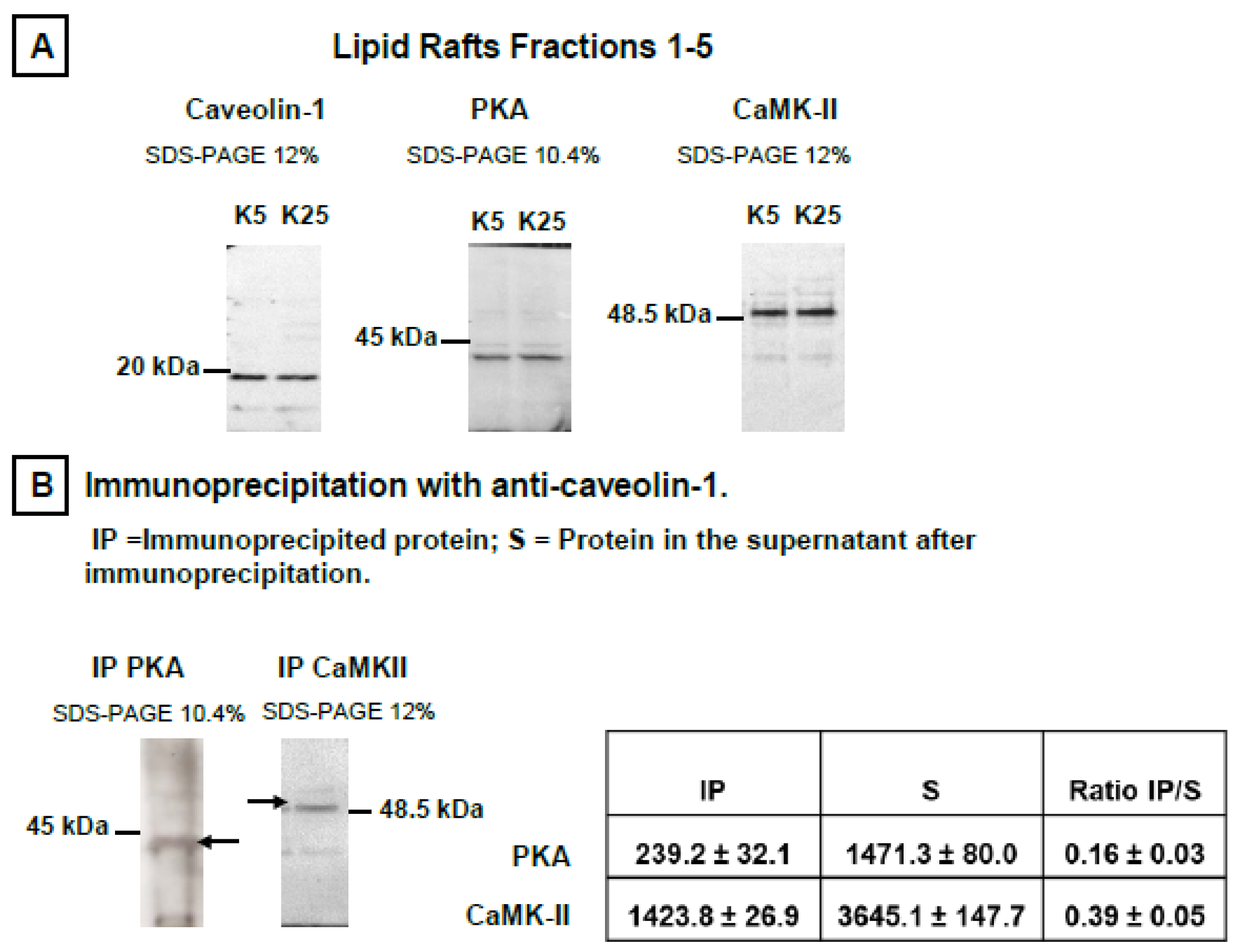
2.2. PKA and CaMK-II Are Associated with Caveolin-1-Rich Lipid Rafts in the Plasma Membrane of CGNs
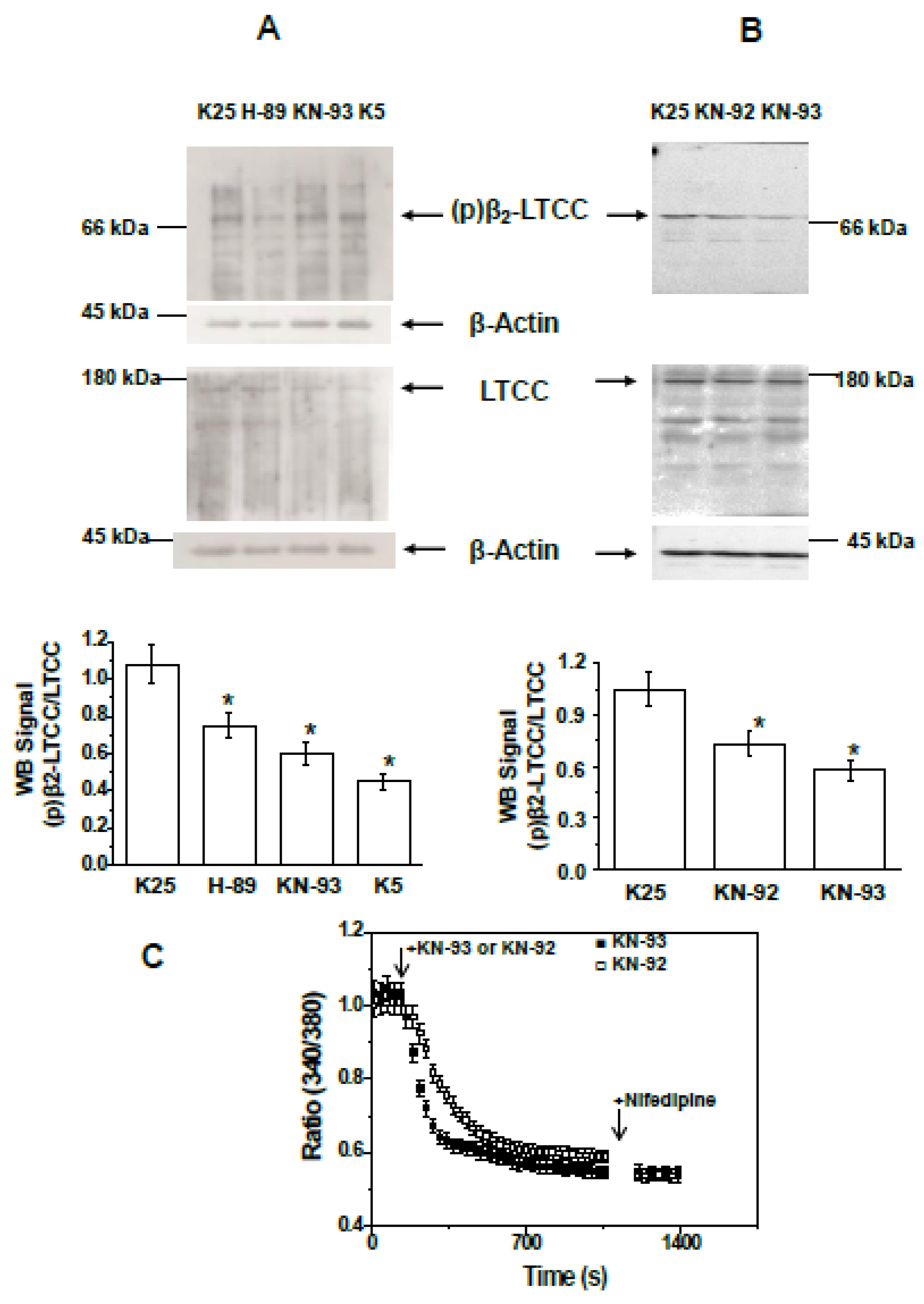
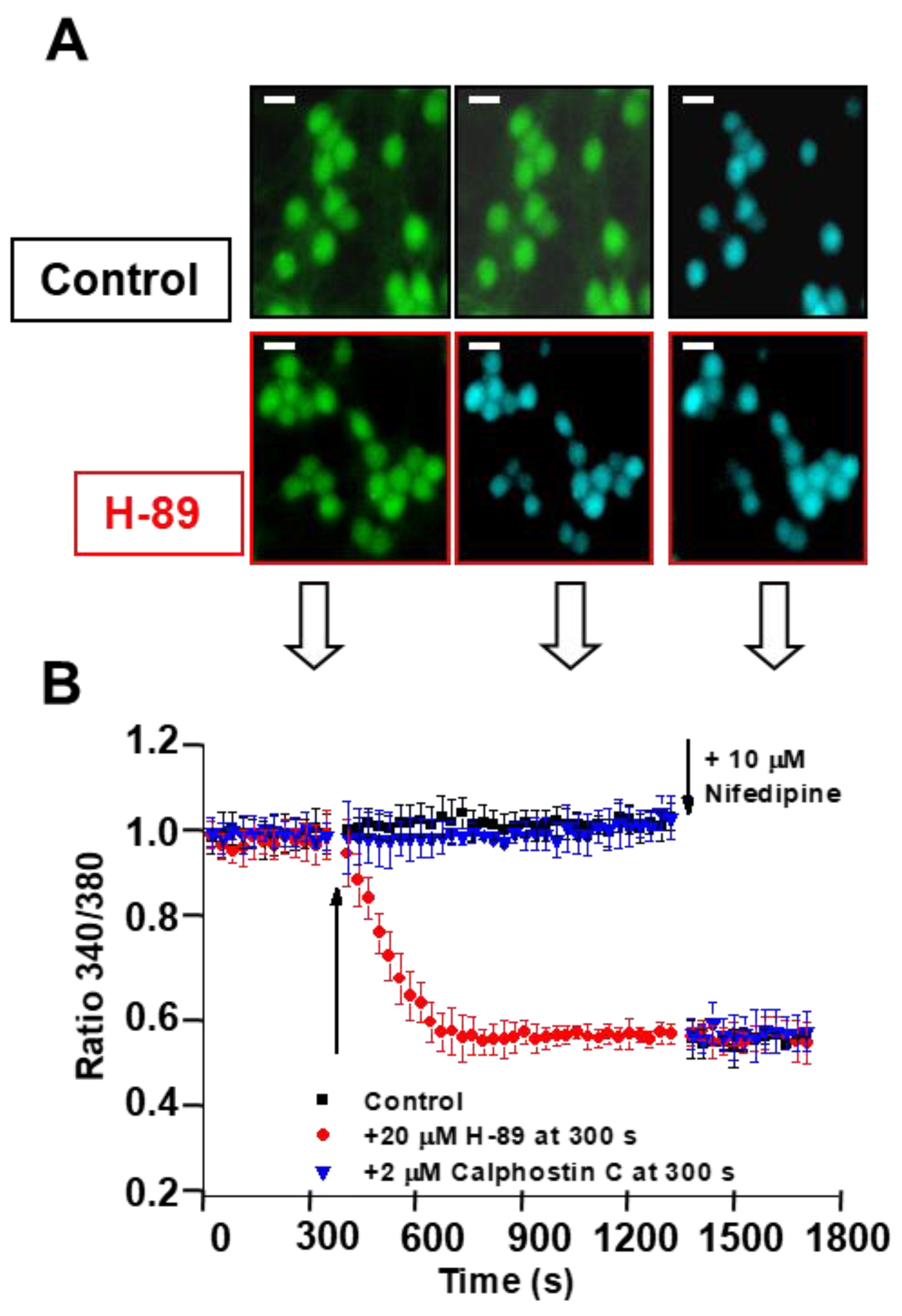
2.3. PKA and CAMK-II Inhibitors Decrease the Phosphorylation Level of the β2 Subunit of LTCCs to the Phosphorylation Levels Measured after CGN Treatment with MβCD and Also in Proapoptotic K5 Medium
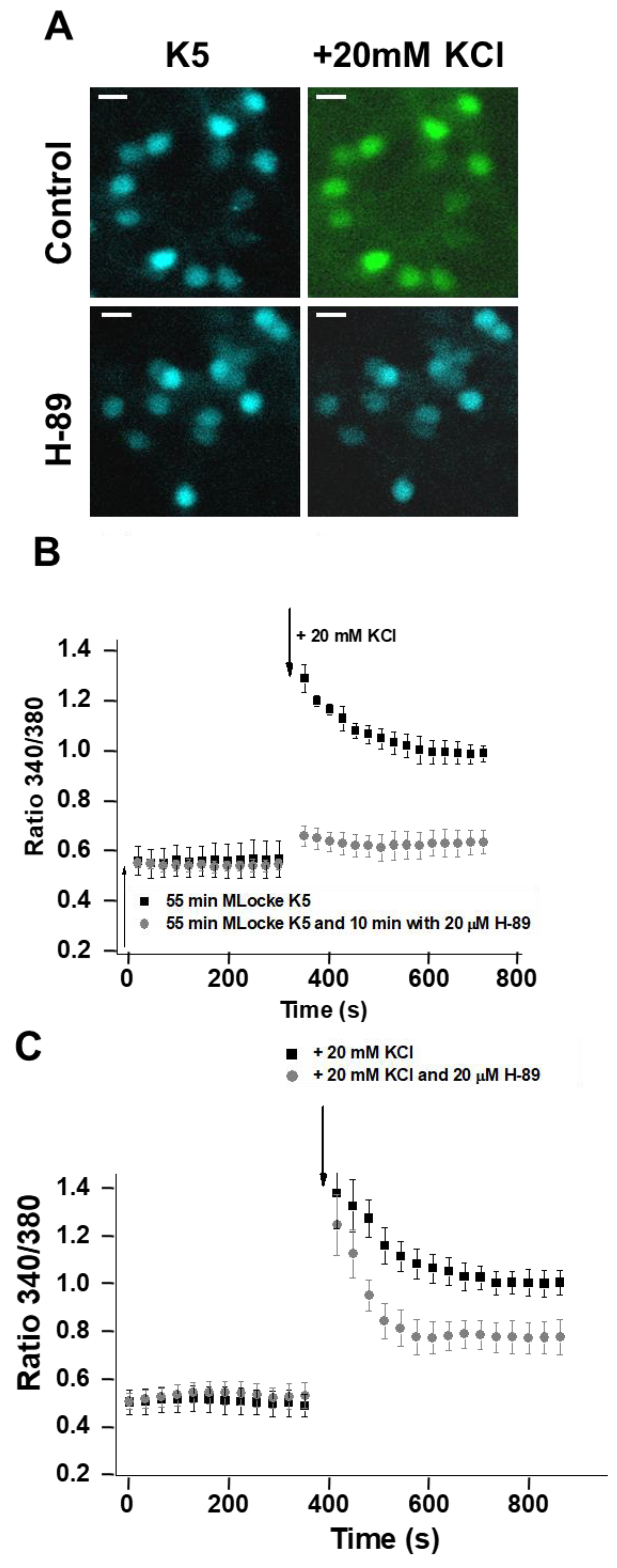
2.4. Treatment of CGNs with MβCD or with the PKA Inhibitor H-89 Lowers Steady-State [Ca2+]i to Values Close to Those Attained in Proapoptotic K5 Medium
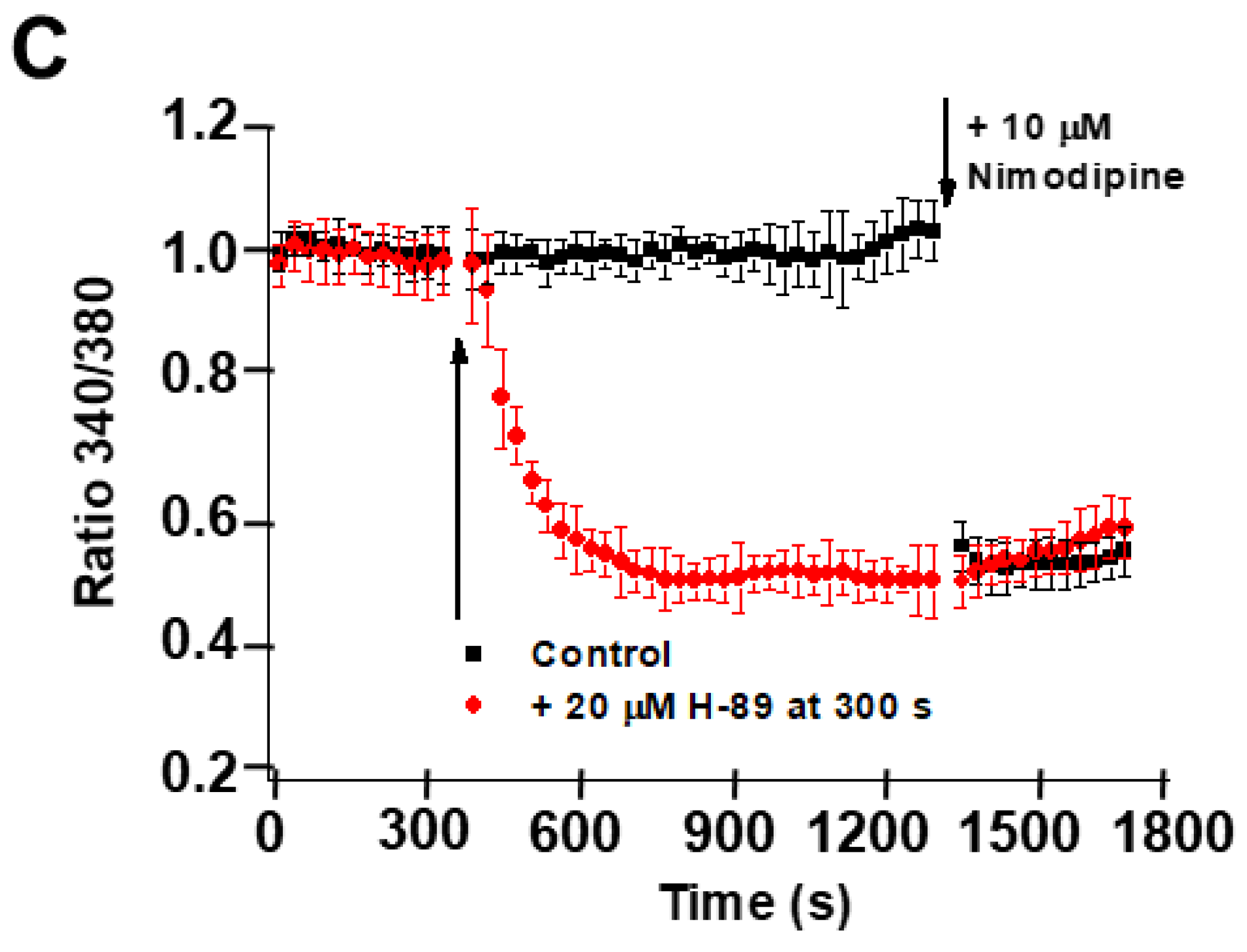
2.5. Inhibition of PKA Alters the LTCC Response to Partial Depolarization of the Plasma Membrane in CGNs
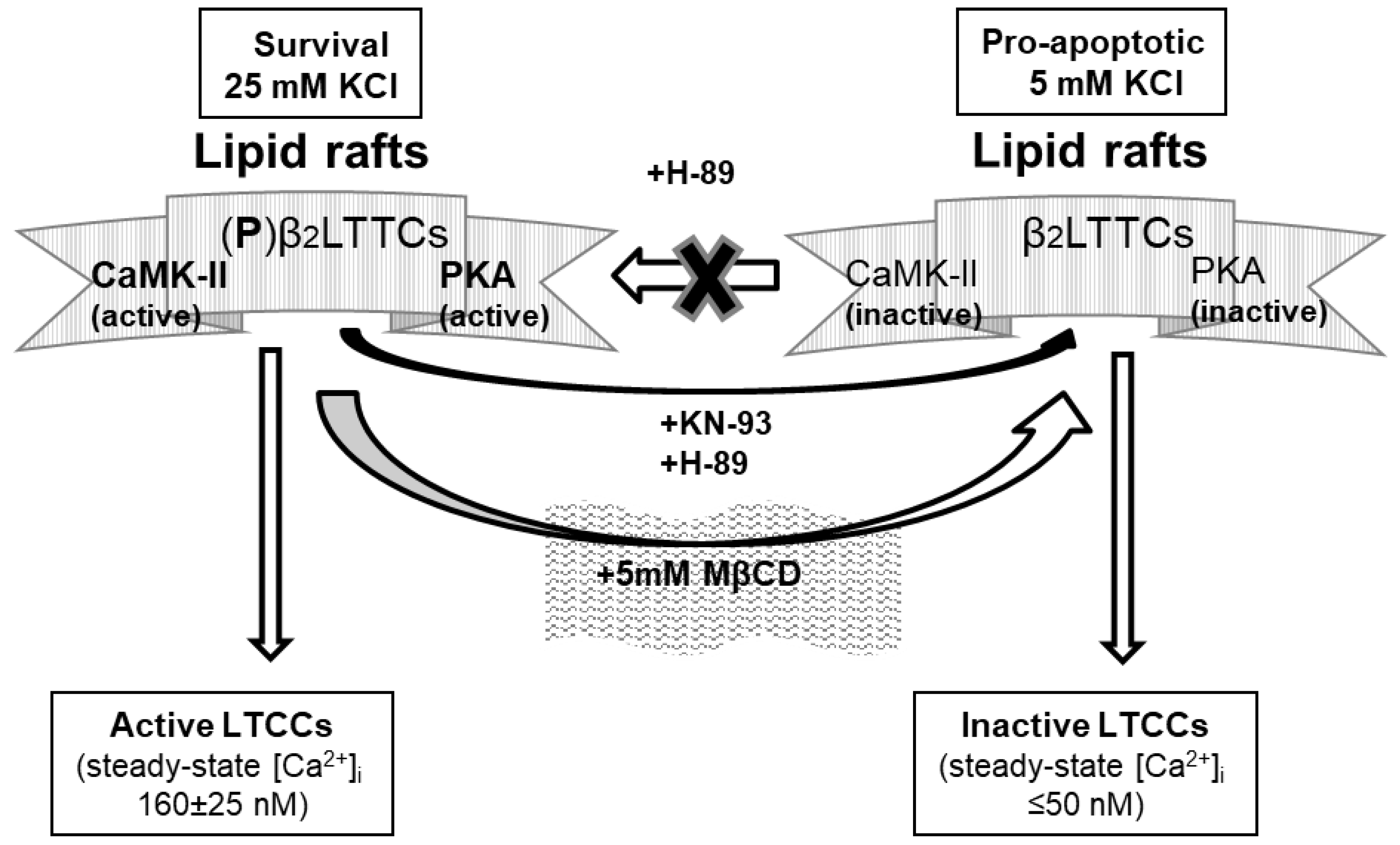
3. Discussion
4. Materials and Methods
4.1. Preparation of Rat Cerebellar Granule Neurons (CGNs)
4.2. Measurement of the Intracellular Free Ca2+ Concentration ([Ca2+]i)
4.3. CGN Cell Lysates and Western Blotting
4.4. Lipid Raft Preparation
4.5. Immunoprecipitation
4.6. Measurement of Cholesterol Content in Cell Lysates
4.7. CGN Treatments
4.8. Chemicals and Reagents
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| [Ca2+]i | intracellular free calcium concentration |
| CaMK-II | calcium/calmodulin-dependent protein kinase II |
| CGN | cerebellar granule neurons |
| DMEM | Dulbecco’s modified Eagle medium |
| EDTA | ethylenediamine tetraacetic acid |
| Fura-2 AM | Fura-2-acetoxymethyl ester |
| LTCC | L-type calcium channels |
| MβCD | methyl-β-cyclodextrin |
| NMDAr, | N-methyl d-aspartate receptor |
| PBS | phosphate-buffered saline |
| PBST | PBS supplemented with 0.05% polyoxyethylenesorbitan monolaurate (Tween 20) |
| PKA | protein kinase A |
| PKC | protein kinase C |
| VGCCs | voltage-gated calcium channels. |
References
- Tiwari, G.; Tiwari, R.; Rai, A.K. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied Sci. 2010, 2, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Shelley, R.H.; Babu, J. Role of Cyclodextrins in Nanoparticle-Based Drug Delivery Systems. J. Pharm. Sci. 2018, 107, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- López-Nicolás, J.M.; Rodríguez-Bonilla, P.; García-Carmona, F. Cyclodextrins and Antioxidants. Crit. Rev. Food Sci. Nutr. 2014, 54, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Crumling, M.A.; King, K.A.; Duncan, R.K. Cyclodextrins and Iatrogenic Hearing Loss: New Drugs with Significant Risk. Front. Cell. Neurosci. 2017, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.; Nayak, D.P. Lipid Raft Disruption by Cholesterol Depletion Enhances Influenza A Virus Budding from MDCK Cells. J. Virol. 2007, 81, 12169–12178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.-C.; Chiang, Y.-T.; Lin, Y.-C.; Chang, Y.-T.; Lu, C.-Y.; Chen, T.-Y.; Yeh, C.-S. Disruption of Lipid Raft Function Increases Expression and Secretion of Monocyte Chemoattractant Protein-1 in 3T3-L1 Adipocytes. PLoS ONE 2016, 11, e0169005. [Google Scholar] [CrossRef] [PubMed]
- Ulloth, J.E.; Almaguel, F.G.; Padilla, A.; Bu, L.; Liu, J.-L.; De Leon, M. Characterization of methyl-β-cyclodextrin toxicity in NGF-differentiated PC12 cell death. NeuroToxicology 2007, 28, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques-da-Silva, D.; Gutierrez-Merino, C. L-type voltage-operated calcium channels, N-methyl-d-aspartate receptors and neuronal nitric-oxide synthase form a calcium/redox nano-transducer within lipid rafts. Biochem. Biophys. Res. Commun. 2012, 420, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Marques-da-Silva, D.; Gutierrez-Merino, C. Caveolin-rich lipid rafts of the plasma membrane of mature cerebellar granule neurons are microcompartments for calcium/reactive oxygen and nitrogen species cross-talk signalling. Cell Calcium 2014, 56, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Marques-da-Silva, D.; Samhan-Arias, A.K.; Tiago, T.; Gutierrez-Merino, C. L-type calcium channels and cytochrome b5 reductase are components of protein complexes tightly associated with lipid rafts microdomains of the neuronal plasma membrane. J. Proteomics 2010, 73, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C. Beta subunits of voltage-gated calcium channels. J. Bioenerg. Biomembr. 2003, 35, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Buraei, Z.; Yang, J. Structure and function of the β subunit of voltage-gated Ca²⁺ channels. Biochim. Biophys. Acta 2013, 1828, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Hohaus, A.; Person, V.; Behlke, J.; Schaper, J.; Morano, I.; Haase, H. The carboxyl-terminal region of AHNAK provides a link between cardiac L-type Ca2+ channels and the actin-based cytoskeleton. FASEB J. 2002, 16, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Tiago, T.; Marques-da-Silva, D.; Samhan-Arias, A.K.; Aureliano, M.; Gutierrez-Merino, C. Early disruption of the actin cytoskeleton in cultured cerebellar granule neurons exposed to 3-morpholinosydnonimine-oxidative stress is linked to alterations of the cytosolic calcium concentration. Cell Calcium 2011, 49, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Rubin, C.S.; Lisanti, M.P. Regulation of cAMP-mediated Signal Transduction via Interaction of Caveolins with the Catalytic Subunit of Protein Kinase A. J. Biol. Chem. 1999, 274, 26353–26360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davare, M.A.; Dong, F.; Rubin, C.S.; Hell, J.W. The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J. Biol. Chem. 1999, 274, 30280–30287. [Google Scholar] [CrossRef] [PubMed]
- Gallo, V.; Kingsbury, A.; Balázs, R.; Jørgensen, O.S. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J. Neurosci. 1987, 7, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Bellone, C.; Cattabeni, F.; Di Luca, M. Protein Kinase C Activation Modulates α-Calmodulin Kinase II Binding to NR2A Subunit of N-Methyl-D-Aspartate Receptor Complex. J. Biol. Chem. 2001, 276, 7609–7613. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, X.; Liu, H.; Cao, Z.; Chen, S.; Cao, B.; Liu, J. Phosphorylated CaMKII post-synaptic binding to NR2B subunits in the anterior cingulate cortex mediates visceral pain in visceral hypersensitive rats. J. Neurochem. 2012, 121, 662–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Du, F.; Tian, Q.-B.; Zhang, J.; Endo, S. Ca2+/calmodulin-dependent protein kinase IIα clusters are associated with stable lipid rafts and their formation traps PSD-95. J. Neurochem. 2008, 10, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Pinard, C.R.; Mascagni, F.; McDonald, A.J. Neuronal localization of Cav1.2 L-type calcium channels in the rat basolateral amygdale. Brain Res. 2005, 1064, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Bünemann, M.; Gerhardstein, B.L.; Gao, T.; Hosey, M.M. Functional Regulation of L-type Calcium Channels via Protein Kinase A-mediated Phosphorylation of the β2 Subunit. J. Biol. Chem. 1999, 274, 33851–33854. [Google Scholar] [CrossRef] [PubMed]
- Kamp, T.J.; Hell, J.W. Regulation of Cardiac L-Type Calcium Channels by Protein Kinase A and Protein Kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudmon, A.; Schulman, H.; Kim, J.; Maltez, J.M.; Tsien, R.W.; Pitt, G.S. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 2005, 171, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Karl, R.; Moosmang, S.; Lenhardt, P.; Klugbauer, N.; Hofmann, F.; Kleppisch, T.; Welling, A. Calmodulin kinase II is involved in voltage dependent facilitation of the L-type Cav1.2 calcium channel: Identification of the phosphorylation sites. J. Biol. Chem. 2006, 281, 25560–25567. [Google Scholar] [CrossRef] [PubMed]
- Grueter, C.E.; Abiria, S.A.; Wu, Y.; Anderson, M.E.; Colbran, R.J. Differential regulated interactions of calcium/calmodulin-dependent protein kinase II with isoforms of voltage-gated calcium channel beta subunits. Biochemistry 2008, 47, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Schengrund, C.-L. Lipid rafts: Keys to neurodegeneration. Brain Res. Bull. 2010, 82, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Sebastião, A.M.; Colino-Oliveira, M.; Assaife-Lopes, N.; Dias, R.B.; Ribeiro, J.A. Lipid rafts, synaptic transmission and plasticity: Impact in age-related neurodegenerative diseases. Neuropharmacology 2013, 64, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Sonnino, S.; Aureli, M.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Lipid rafts in neurodegeneration and neuroprotection. Mol. Neurobiol. 2014, 50, 130–148. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Kim, T.-W. Linking Lipids to Alzheimer’s Disease: Cholesterol and Beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J.L.; Johnson, E.M., Jr. Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992, 15, 501–508. [Google Scholar] [CrossRef]
- Gutierrez-Martin, Y.; Martin-Romero, F.J.; Henao, F.; Gutierrez-Merino, C. Alteration of cytosolic free calcium homeostasis by SIN-1: High sensitivity of L-type Ca2+ channels to extracellular oxidative/nitrosative stress in cerebellar granule cells. J. Neurochem. 2005, 92, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bereguiain, M.A.; Samhan-Arias, A.K.; Martin-Romero, F.J.; Gutierrez-Merino, C. Hydrogen sulfide raises cytosolic calcium in neurons through activation of L-type Ca2+ channels. Antioxid. Redox Signal. 2008, 10, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Usai, C. High affinity block by nimodipine of the internal calcium elevation in chronically depolarized rat cerebellar granule neurons. Neurosci. Lett. 1996, 207, 77–80. [Google Scholar] [CrossRef]
- Evans, G.J.; Pocock, J.M. Modulation of neurotransmitter release by dihydropyridine-sensitive calcium channels involves tyrosine phosphorylation. Eur. J. Neurosci. 1999, 11, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Toescu, E.C. Activity of voltage-operated calcium channels in rat cerebellar granule neurons and neuronal survival. Neuroscience 1999, 94, 561–570. [Google Scholar] [CrossRef]
- D’Mello, S.R.; Galli, C.; Ciotti, T.; Calissano, P. Induction of apoptosis in cerebellar granule neurons by low potassium: Inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA 1993, 90, 10989–10993. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.Y.; Korolev, V.V.; Wang, J.Z. Cyclic AMP and pituitary adenylate cyclase-activating polypeptide (PACAP) prevent programmed cell death of cultured cerebellar granule cells. Neurosci. Lett. 1996, 206, 181–184. [Google Scholar] [CrossRef]
- Campard, P.K.; Crochemore, C.; Rene, F.; Monnier, D.; Koch, B.; Loefer, J.P. PACAP type I receptor activation promotes cerebellar neuron survival through the cAMP/PKA signaling pathway. DNA Cell Biol. 1997, 16, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 2002, 1, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Tokumitsu, H.; Soderling, T.R. Calcium promotes cell survival through CaM-kinase activation of the protein-kinase B pathway. Nature 1998, 396, 584–587. [Google Scholar] [CrossRef] [PubMed]
- See, V.; Boutillier, A.R.; Bito, H.; Loefer, J.P. Calcium/calmodulin-dependent protein kinase IV (CaMKIV) inhibits apoptosis induced by potassium deprivation in cerebellar granule neurons. FASEB J. 2001, 15, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Merino, C.; Marques-da-Silva, D.; Fortalezas, S.; Samhan-Arias, A.K. The critical role of lipid rafts nanodomains in the cross-talk between calcium and reactive oxygen and nitrogen species in cerebellar granule neurons apoptosis by extracellular potassium deprivation. AIMS Mol. Sci. 2016, 3, 12–29. [Google Scholar] [CrossRef]
- Anderson, M.E.; Braun, A.P.; Wu, Y.; Lu, T.; Wu, Y.; Schulman, H.; Sung, R.J. KN-93, an Inhibitor of Multifunctional Ca++/Calmodulin-Dependent Protein Kinase, Decreases Early After Depolarizations in Rabbit Heart. J. Pharm. Exp. Ther. 1998, 287, 996–1006. [Google Scholar]
- Samhan-Arias, A.K.; Martin-Romero, F.J.; Gutierrez-Merino, C. Kaempferol blocks oxidative stress in cerebellar granule cells and reveals a key role for the plasma membrane NADH oxidase activity in the commitment of apoptosis. Free Radic. Biol. Med. 2004, 37, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Martin-Romero, F.J.; Garcia-Martin, E.; Gutierrez-Merino, C. Inhibition of the oxidative stress produced by plasma membrane NADH oxidase delays low-potassium induced apoptosis of cerebellar granule cells. J. Neurochem. 2002, 82, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Samhan-Arias, A.K.; Marques-da-Silva, D.; Yanamala, N.; Gutierrez-Merino, C. Stimulation and clustering of cytochrome b5 reductase in caveolin-rich lipid microdomains is an early event in oxidative stress-mediated apoptosis of cerebellar granule neurons. J. Proteomics 2012, 75, 2934–2949. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Bayer, K.U. CaMKII regulation in information processing and storage. Trends Neurosci. 2012, 35, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, R.P.; Cheng, H.; Lederer, W.J.; Suzuki, T.; Lakatta, E.G. Dual regulation of Ca2+/calmodulin-dependent kinase II activity by membrane voltage and by calcium influx. Proc. Natl. Acad. Sci. USA 1994, 91, 9659–9663. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, H.; Song, Y.; Watanabe, M.; Masumiya, H.; Gupte, S.A.; Ochi, R.; Okada, T. Cholesterol depletion modulates basal L-type Ca2+ current and abolishes its β-adrenergic enhancement in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H285–H292. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.C.; Nagano, N.; Kuenzi, F.M.; Jarolimek, W.; Huber, I.; Walter, D.; Wietzorrek, G.; Boyce, S.; Kullmann, D.M.; Striessnig, J.; et al. Neurological phenotype and synaptic function in mice lacking the CaV1.3 alpha subunit of neuronal L-type voltage dependent Ca2+ channels. Neuroscience 2003, 120, 435–442. [Google Scholar] [CrossRef]
- Koschak, A.; Obermair, G.J.; Pivotto, F.; Sinnegger-Brauns, M.J.; Striessnig, J.; Pietrobon, D. Molecular nature of anomalous L-type calcium channels in mouse cerebellar granule cells. J. Neurosci. 2007, 27, 3855–3863. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condorius, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Chen, B.; Hund, T.J.; Koval, O.M.; Purohit, A.; Song, L.S.; Mohler, P.J.; Anderson, M.E. Proarrhythmic defects in Timothy syndrome require calmodulin kinase II. Circulation 2008, 118, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Day, M.; Wang, Z.; Ding, J.; An, X.; Ingham, C.A.; Shering, A.F.; Wokosin, D.; Ilijic, E.; Sun, Z.; Sampson, A.R.; et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 2006, 9, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Moosmang, S.; Haider, N.; Klugbauer, N.; Adelsberger, H.; Langwieser, N.; Müller, J.; Stiess, M.; Marais, E.; Schulla, V.; Lacinova, L.; et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J. Neurosci. 2005, 25, 9883–9892. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Flockerzi, V.; Hofmann, F. Regional Expression and Cellular Localization of the α1 and β Subunit of High Voltage-Activated Calcium Channels in Rat Brain. J. Neurosci. 1997, 17, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Abiria, S.A.; Colbran, R.J. CaMKII associates with CaV1.2 L-type calcium channels via selected β subunits to enhance regulatory phosphorylation. J. Neurosci. 2010, 112, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Soltis, A.R.; Saucerman, J.J. Synergy between CaMKII Substrates and β-Adrenergic Signaling in Regulation of Cardiac Myocyte Ca2+ Handling. Biophys. J. 2010, 99, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Samhan-Arias, A.K.; Garcia-Bereguiain, M.A.; Martin-Romero, F.J.; Gutierrez-Merino, C. Clustering of plasma membrane-bound cytochrome b5 reductase within ‘lipid rafts’ microdomains of the neuronal plasma membrane. Mol. Cell. Neurosci. 2009, 40, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Fortalezas, S.; Marques-da-Silva, D.; Gutierrez-Merino, C. Creatine protects against cytosolic calcium dysregulation, mitochondrial depolarization and increase of reactive oxygen species production in rotenone-induced cell death of cerebellar granule neurons. Neurotox. Res. 2018, 34, 717–732. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [MβCD] (mM) | Cholesterol Content (nmol/mg of CGN Protein) | Cell Viability |
|---|---|---|
| 0 | 22.8 ± 2.1 | 100 ± 5% |
| 5 | 8.4 ± 0.9 | 100 ± 5% |
| 10 | 4.6 ± 0.4 | 100 ± 6% |
| 20 | 4.7 ± 0.4 | 95 ± 5% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fortalezas, S.; Marques-da-Silva, D.; Gutierrez-Merino, C. Methyl-β-Cyclodextrin Impairs the Phosphorylation of the β2 Subunit of L-Type Calcium Channels and Cytosolic Calcium Homeostasis in Mature Cerebellar Granule Neurons. Int. J. Mol. Sci. 2018, 19, 3667. https://doi.org/10.3390/ijms19113667
Fortalezas S, Marques-da-Silva D, Gutierrez-Merino C. Methyl-β-Cyclodextrin Impairs the Phosphorylation of the β2 Subunit of L-Type Calcium Channels and Cytosolic Calcium Homeostasis in Mature Cerebellar Granule Neurons. International Journal of Molecular Sciences. 2018; 19(11):3667. https://doi.org/10.3390/ijms19113667
Chicago/Turabian StyleFortalezas, Sofia, Dorinda Marques-da-Silva, and Carlos Gutierrez-Merino. 2018. "Methyl-β-Cyclodextrin Impairs the Phosphorylation of the β2 Subunit of L-Type Calcium Channels and Cytosolic Calcium Homeostasis in Mature Cerebellar Granule Neurons" International Journal of Molecular Sciences 19, no. 11: 3667. https://doi.org/10.3390/ijms19113667
APA StyleFortalezas, S., Marques-da-Silva, D., & Gutierrez-Merino, C. (2018). Methyl-β-Cyclodextrin Impairs the Phosphorylation of the β2 Subunit of L-Type Calcium Channels and Cytosolic Calcium Homeostasis in Mature Cerebellar Granule Neurons. International Journal of Molecular Sciences, 19(11), 3667. https://doi.org/10.3390/ijms19113667