Targeting Smoothened as a New Frontier in the Functional Recovery of Central Nervous System Demyelinating Pathologies



Abstract
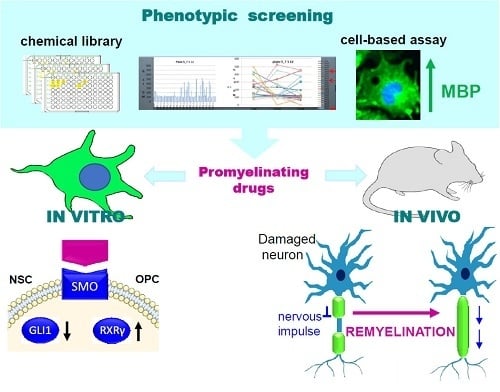
:
{kind=link}
{kind=link}
1. Introduction
2. Shh Signaling during CNS Remyelination
3. Shh/Smo Signaling During OPC Differentiation
4. Smo and Gli-Associated Oncogene Regulation in Adult Somatic Cells
4.1. Smo and Gli Antagonists
4.2. Canonical Pathways of Gli Regulation
4.3. Non-Canonical Pathways of Gli Regulation
5. Shh Signaling, Cholesterol Biosynthesis and Myelination: A Complex Liaison
6. Concluding Remarks
Acknowledgments
Conflict of interest
References
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and Mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.-W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, D.; Stone, D.M.; Brush, J.; Ryan, A.; Armanini, M.; Frantz, G.; Rosenthal, A.; De Sauvage, F.J. Characterization of Two Patched Receptors for the Vertebrate Hedgehog Protein Family. Proc. Natl. Acad. Sci. USA 1998, 95, 13630–13634. [Google Scholar] [CrossRef] [PubMed]
- Buglino, J.A.; Resh, M.D. Hhat Is a Palmitoylacyltransferase with Specificity for N-Palmitoylation of Sonic Hedgehog. J. Biol. Chem. 2008, 283, 22076–22088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferent, J.; Zimmer, C.; Durbec, P.; Ruat, M.; Traiffort, E. Sonic Hedgehog Signaling Is a Positive Oligodendrocyte Regulator during Demyelination. J. Neurosci. 2013, 33, 1759–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA Interaction Is a Druggable Target for Hedgehog-Dependent Tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Chew, L.-J.; DeBoy, C.A. Pharmacological Approaches to Intervention in Hypomyelinating and Demyelinating White Matter Pathology. Neuropharmacology 2016, 110, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Kremer, D.; Göttle, P.; Hartung, H.-P.; Küry, P. Pushing Forward: Remyelination as the New Frontier in CNS Diseases. Trends Neurosci. 2016, 39, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Villoslada, P. Neuroprotective Therapies for Multiple Sclerosis and Other Demyelinating Diseases. Mult. Scler. Demyelinating Disord. 2016, 1, 1. [Google Scholar] [CrossRef]
- Joubert, L.; Foucault, I.; Sagot, Y.; Bernasconi, L.; Duval, F.; Alliod, C.; Frossard, M.-J.; Pescini Gobert, R.; Curchod, M.-L.; Salvat, C.; et al. Chemical Inducers and Transcriptional Markers of Oligodendrocyte Differentiation. J. Neurosci. Res. 2010, 88, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A Regenerative Approach to the Treatment of Multiple Sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-Based Modulation of Endogenous Stem Cells Promotes Functional Remyelination In Vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Porcu, G.; Serone, E.; De Nardis, V.; Di Giandomenico, D.; Lucisano, G.; Scardapane, M.; Poma, A.; Ragnini-Wilson, A. Clobetasol and Halcinonide Act as Smoothened Agonists to Promote Myelin Gene Expression and RxRγ Receptor Activation. PLoS ONE 2015, 10, e0144550. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Su, T.; Verkman, A.S. Clobetasol Promotes Remyelination in a Mouse Model of Neuromyelitis Optica. Acta Neuropathol. Commun. 2016, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Hubler, Z.; Allimuthu, D.; Bederman, I.; Elitt, M.S.; Madhavan, M.; Allan, K.C.; Shick, H.E.; Garrison, E.; Karl, M.; Factor, D.C.; et al. Accumulation of 8,9-Unsaturated Sterols Drives Oligodendrocyte Formation and Remyelination. Nature 2018, 560, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Ashikawa, Y.; Nishimura, Y.; Okabe, S.; Sasagawa, S.; Murakami, S.; Yuge, M.; Kawaguchi, K.; Kawase, R.; Tanaka, T. Activation of Sterol Regulatory Element Binding Factors by Fenofibrate and Gemfibrozil Stimulates Myelination in Zebrafish. Front. Pharmacol. 2016, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, G.; Sircar, R.; Kong, J.H.; Nachtergaele, S.; Sagner, A.; Byrne, E.F.; Covey, D.F.; Siebold, C.; Rohatgi, R. Cholesterol Activates the G-Protein Coupled Receptor Smoothened to Promote Hedgehog Signaling. eLife 2016, 5, e20304. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zheng, S.; Wierbowski, B.M.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 2018, 174, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, J.; Bond, M.C.; Chen, M.; Ren, X.-R.; Lyerly, H.K.; Barak, L.S. Identification of Select Glucocorticoids as Smoothened Agonists: Potential Utility for Regenerative Medicine. Proc. Natl. Acad. Sci. USA 2010, 107, 9323–9328. [Google Scholar] [CrossRef] [PubMed]
- Mangelberger, D.; Kern, D.; Loipetzberger, A.; Eberl, M.; Aberger, F. Cooperative Hedgehog-EGFR Signaling. Front. Biosci. 2012, 17, 90–99. [Google Scholar] [CrossRef]
- Dirix, L. Discovery and Exploitation of Novel Targets by Approved Drugs. J. Clin. Oncol. 2014, 32, 720–721. [Google Scholar] [CrossRef] [PubMed]
- Gibney, S.M.; McDermott, K.W. Sonic hedgehog promotes the generation of myelin proteins by transplanted oligosphere-derived cells. J. Neurosci. Res. 2009, 87, 3067–3075. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Jay, P.; Berta, P.; Scherer, S.W.; Tsui, L.C.; Muenke, M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 1996, 14, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Bertolacini, C.D.; Richieri-Costa, A.; Ribeiro-Bicudo, L.A. Sonic hedgehog (SHH) mutation in patients within the spectrum of holoprosencephaly. Brain Dev. 2010, 32, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Deoni, S.C.L.; Mercure, E.; Blasi, A.; Gasston, D.; Thomson, A.; Johnson, M.; Williams, S.C.R.; Murphy, D.G.M. Mapping Infant Brain Myelination with Magnetic Resonance Imaging. J. Neurosci. 2011, 31, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Young, K.M. White Matter Plasticity in Adulthood. Neuroscience 2014, 276, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.G.; Lyons, D.A. On Myelinated Axon Plasticity and Neuronal Circuit Formation and Function. J. Neurosci. 2017, 37, 10023–10034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auer, F.; Vagionitis, S.; Czopka, T. Evidence for Myelin Sheath Remodeling in the CNS Revealed by In Vivo Imaging. Curr. Biol. 2018, 28, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Tomassy, G.S.; Dershowitz, L.B.; Arlotta, P. Diversity Matters: A Revised Guide to Myelination. Trends Cell Biol. 2016, 26, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaller, M.S.; Lazari, A.; Blanco-Duque, C.; Sampaio-Baptista, C.; Johansen-Berg, H. Myelin Plasticity and Behaviour-Connecting the Dots. Curr. Opin. Neurobiol. 2017, 47, 86–92. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, I.A.; Ohayon, D.; Li, H.; De Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor Skill Learning Requires Active Central Myelination. Science 2014, 346, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Ihrie, R.A.; Shah, J.K.; Harwell, C.C.; Levine, J.H.; Guinto, C.D.; Lezameta, M.; Kriegstein, A.R.; Alvarez-Buylla, A. Persistent Sonic Hedgehog Signaling in Adult Brain Determines Neural Stem Cell Positional Identity. Neuron 2011, 71, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferent, J.; Cochard, L.; Faure, H.; Taddei, M.; Hahn, H.; Ruat, M.; Traiffort, E. Genetic Activation of Hedgehog Signaling Unbalances the Rate of Neural Stem Cell Renewal by Increasing Symmetric Divisions. Stem Cell Rep. 2014, 3, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, E.; Arlotta, P. The Quest for Myelin in the Adult Brain. Nat. Cell Biol. 2013, 15, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Daynac, M.; Pineda, J.R.; Chicheportiche, A.; Gauthier, L.R.; Morizur, L.; Boussin, F.D.; Mouthon, M.A. TGFβ lengthens the G1 phase of stem cells in aged mouse brain. Stem Cells 2014, 32, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Tekki-Kessaris, N.; Woodruff, R.; Hall, A.C.; Gaffield, W.; Kimura, S.; Stiles, C.D.; Rowitch, D.H.; Richardson, W.D. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development 2001, 128, 2545–2554. [Google Scholar] [PubMed]
- Lai, K.; Kaspar, B.K.; Gage, F.H.; Schaffer, D.V. Sonic Hedgehog Regulates Adult Neural Progenitor Proliferation in Vitro and In Vivo. Nat. Neurosci. 2003, 6, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Joyner, A.L. In Vivo Analysis of Quiescent Adult Neural Stem Cells Responding to Sonic Hedgehog. Nature 2005, 437, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Zeisel, A.; Codeluppi, S.; Van Bruggen, D.; Mendanha Falcão, A.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte Heterogeneity in the Mouse Juvenile and Adult Central Nervous System. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Charytoniuk, D.; Traiffort, E.; Hantraye, P.; Hermel, J.M.; Galdes, A.; Ruat, M. Intrastriatal Sonic Hedgehog Injection Increases Patched Transcript Levels in the Adult Rat Subventricular Zone. Eur. J. Neurosci. 2002, 16, 2351–2357. [Google Scholar] [CrossRef] [PubMed]
- Machold, R.; Hayashi, S.; Rutlin, M.; Muzumdar, M.D.; Nery, S.; Corbin, J.G.; Gritli-Linde, A.; Dellovade, T.; Porter, J.A.; Rubin, L.L.; et al. Sonic Hedgehog Is Required for Progenitor Cell Maintenance in Telencephalic Stem Cell Niches. Neuron 2003, 39, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Huang, S.; Yang, L.; Zhao, L.; Yin, Y.; Liu, Z.; Chen, Z.; Zhang, H. Down-Regulation of Sonic Hedgehog Signaling Pathway Activity Is Involved in 5-Fluorouracil-Induced Apoptosis and Motility Inhibition in Hep3B Cells. Acta Biochim. Biophys. Sin. 2008, 40, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Sirko, S.; Behrendt, G.; Johansson, P.A.; Tripathi, P.; Costa, M.; Bek, S.; Heinrich, C.; Tiedt, S.; Colak, D. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. Cell Stem Cell 2013, 12, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Harsan, L.-A.; Steibel, J.; Zaremba, A.; Agin, A.; Sapin, R.; Poulet, P.; Guignard, B.; Parizel, N.; Grucker, D.; Boehm, N.; et al. Recovery from Chronic Demyelination by Thyroid Hormone Therapy: Myelinogenesis Induction and Assessment by Diffusion Tensor Magnetic Resonance Imaging. J. Neurosci. 2008, 28, 14189–14201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loulier, K.; Ruat, M.; Traiffort, E. Increase of Proliferating Oligodendroglial Progenitors in the Adult Mouse Brain upon Sonic Hedgehog Delivery in the Lateral Ventricle. J. Neurochem. 2006, 98, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Wegener, A.; Deboux, C.; Bachelin, C.; Frah, M.; Kerninon, C.; Seilhean, D.; Weider, M.; Wegner, M.; Nait-Oumesmar, B. Gain of Olig2 Function in Oligodendrocyte Progenitors Promotes Remyelination. Brain 2015, 138 Pt 1, 120–135. [Google Scholar] [CrossRef]
- Angot, E.; Loulier, K.; Nguyen-Ba-Charvet, K.T.; Gadeau, A.-P.; Ruat, M.; Traiffort, E. Chemoattractive Activity of Sonic Hedgehog in the Adult Subventricular Zone Modulates the Number of Neural Precursors Reaching the Olfactory Bulb. Stem Cells 2008, 26, 2311–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breachbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Goderie, S.K.; Temple, S. Asymmetric distribution of EGFR receptor during mitosis generates diverse CNS progenitor cells. Neuron 2005, 45, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Samanta, J.; Grund, E.M.; Silva, H.M.; Lafaille, J.J.; Fishell, G.; Salzer, J.L. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature 2015, 526, 448. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The Mechanisms of Hedgehog Signaling and Its Roles in Development and Disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Mierzwa, A.J.; Sullivan, G.M.; Beer, L.A.; Ahn, S.; Armstrong, R.C. Comparison of cortical and white matter traumatic brain injury models reveals differential effects in the subventricular zone and divergent Sonic hedgehog signaling pathways in neuroblasts and oligodendrocyte progenitors. ASN Neuro 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.A.; Armstrong, R.C. Postnatal Sonic Hedgehog (Shh) Responsive Cells Give Rise to Oligodendrocyte Lineage Cells during Myelination and in Adulthood Contribute to Remyelination. Exp. Neurol. 2018, 299, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Gorojankina, T.; Hoch, L.; Faure, H.; Roudaut, H.; Traiffort, E.; Schoenfelder, A.; Girard, N.; Mann, A.; Manetti, F.; Solinas, A.; et al. Discovery, Molecular and Pharmacological Characterization of GSA-10, a Novel Small-Molecule Positive Modulator of Smoothened. Mol. Pharmacol. 2013, 83, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-C.; Almazan, G. Role of Sonic Hedgehog Signaling in Oligodendrocyte Differentiation. Neurochem. Res. 2016, 41, 3289–3299. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Klein, R.S. Mediators of Oligodendrocyte Differentiation during Remyelination. FEBS Lett. 2011, 585, 3730–3737. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.; Sánchez, P.; Schmitt, S.; Snaidero, N.; Mitkovski, M.; Velte, C.; Brückner, B.R.; Alexopoulos, I.; Czopka, T.; Jung, S.Y.; et al. Actin Filament Turnover Drives Leading Edge Growth during Myelin Sheath Formation in the Central Nervous System. Dev. Cell 2015, 34, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Fu, M.-M.; Sloan, S.A.; Ibrahim, A.; Olson, A.; Zaremba, A.; Dugas, J.C.; Wienbar, S.; Caprariello, A.V.; Kantor, C.; et al. CNS Myelin Wrapping Is Driven by Actin Disassembly. Dev. Cell 2015, 34, 152–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechler, M.E.; Byrne, L. CNS Myelin Sheath Lengths Are an Intrinsic Property of Oligodendrocytes. Curr. Biol. 2015, 25, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Leach, M.K.; Redmond, S.A.; Chong, S.Y.C.; Mellon, S.H.; Tuck, S.J.; Feng, Z.-Q.; Corey, J.M.; Chan, J.R. A Culture System to Study Oligodendrocyte Myelination Processes Using Engineered Nanofibers. Nat. Methods 2012, 9, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Emery, B.; Agalliu, D.; Cahoy, J.D.; Watkins, T.A.; Dugas, J.C.; Mulinyawe, S.B.; Ibrahim, A.; Ligon, K.L.; Rowitch, D.H.; Barres, B.A. Myelin Gene Regulatory Factor Is a Critical Transcriptional Regulator Required for CNS Myelination. Cell 2009, 138, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, G.J.; Plemel, J.R.; Assinck, P.; Manesh, S.B.; Muir, F.G.W.; Hirata, R.; Berson, M.; Liu, J.; Wegner, M.; Emery, B. Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol. 2017, 134, 403–422. [Google Scholar] [CrossRef] [PubMed]
- Giner, X.C.; Cotnoir-White, D.; Mader, S.; Lévesque, D. Selective Ligand Activity at Nur/Retinoid X Receptor Complexes Revealed by Dimer-Specific Bioluminescence Resonance Energy Transfer-Based Sensors. FASEB J. 2015, 29, 4256–4267. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.K.; Jarjour, A.A.; Nait Oumesmar, B.; Kerninon, C.; Williams, A.; Krezel, W.; Kagechika, H.; Bauer, J.; Zhao, C.; Baron-Van Evercooren, A.; et al. Retinoid X Receptor Gamma Signaling Accelerates CNS Remyelination. Nat. Neurosci. 2011, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; De Tribolet, N.; Radovanovic, I.; I Altaba, A.R. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2017, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.W.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, E.; Milani, M. Role and inhibition of GLI1 protein in cancer. Lung Cancer Targets Ther. 2018, 9, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetsky, M.; Zhang, X.M.; Bottega, S.; Guicherit, O.; Wichterle, H.; Dudek, H.; Bumcrot, D.; Wang, F.Y.; Jones, S.; Shulok, J.; et al. Small-Molecule Modulators of Hedgehog Signaling: Identification and Characterization of Smoothened Agonists and Antagonists. J. Biol. 2002, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.-P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the Human Smoothened Receptor 7TM Bound to an Antitumor Agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-a Potent Inhibitor of the Hedgehog Pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Nachtergaele, S.; Whalen, D.M.; Mydock, L.K.; Zhao, Z.; Malinauskas, T.; Krishnan, K.; Ingham, P.W.; Covey, D.F.; Siebold, C.; Rohatgi, R. Structure and Function of the Smoothened Extracellular Domain in Vertebrate Hedgehog Signaling. eLife 2013, 2, e01340. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog Signaling by Direct Binding of Cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and Arsenic Trioxide Inhibit Hedgehog Pathway Activation and Tumor Growth Associated with Acquired Resistance to Smoothened Antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.K.; Taipale, J.; Young, K.E.; Maiti, T.; Beachy, P.A. Small Molecule Modulation of Smoothened Activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.B.; Joyner, A.L. Gli1 Can Rescue the In Vivo Function of Gli2. Development 2001, 128, 5161–5172. [Google Scholar] [PubMed]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; Ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of Sonic Hedgehog Mediators by Transforming Growth Factor-Beta: Smad3-Dependent Activation of Gli2 and Gli1 Expression In Vitro and In Vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Falcón-Urrutia, P.; Carrasco, C.M.; Lois, P.; Palma, V.; Roth, A.D. Shh Signaling through the Primary Cilium Modulates Rat Oligodendrocyte Differentiation. PLoS ONE 2015, 10, e0133567. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.L.; Finta, C.; Karlström, M.; Jin, Q.; Schwend, T.; Astorga-Wells, J.; Zubarev, R.A.; Del Campo, M.; Criswell, A.R.; De Sanctis, D.; et al. Structural basis of SUFU-GLI interaction in human Hedgehog signaling regulation. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2563–2579. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, H.M.; Sternheim, N.; Lyons, D.A.; Diamond, B.; Hawkins, T.A.; Woods, I.G.; Bhatt, D.H.; Franzini-Armstrong, C.; Dominguez, C.; Arana, N.; et al. A Genetic Screen Identifies Genes Essential for Development of Myelinated Axons in Zebrafish. Dev. Biol. 2006, 298, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.B.; Stephen, D.; Joyner, A.L. All Mouse Ventral Spinal Cord Patterning by Hedgehog Is Gli Dependent and Involves an Activator Function of Gli3. Dev. Cell 2004, 6, 103–115. [Google Scholar] [CrossRef]
- Park, H.L.; Bai, C.; Platt, K.A.; Matise, M.P.; Beeghly, A.; Hui, C.C.; Nakashima, M.; Joyner, A.L. Mouse GLI1 mutants are viable but have defects in SHH signaling in combination with a GLI2 mutation. Development 2000, 127, 1593–1605. [Google Scholar] [PubMed]
- Kitaura, Y.; Hojo, H.; Komiyama, Y.; Takato, T.; Chung, U.I.; Ohba, S.; Marie, P.J. GLI1 haploinsufficiency leads to decreased bone mass with an uncoupling of bone metabolismin adult mice. PLoS ONE 2014, 9, e109597. [Google Scholar] [CrossRef] [PubMed]
- Palencia-Campos, A.; Ullah, A.; Nevado, J.; Yildirim, R.; Unal, E.; Ciorraga, M.; Barruz, P.; Chico, L.; Piceci-Sparascio, F.; Guida, V.; et al. GLI1 Inactivation Is Associated with Developmental Phenotypes Overlapping with Ellis-van Creveld Syndrome. Hum. Mol. Genet. 2017, 26, 4556–4571. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic Hedgehog Signaling Regulates Gli2 Transcriptional Activity by Suppressing Its Processing and Degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, C.; Wang, B. Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev Biol. 2009, 326, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Gessler, M.; Grzeschik, K.H. GLI3 Zinc-Finger Gene Interrupted by Translocations in Greig Syndrome Families. Nature 1991, 352, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.; Kalff-Suske, M.; Vortkamp, A.; Bornholdt, D.; König, R.; Grzeschik, K.H. Point Mutations in Human GLI3 Cause Greig Syndrome. Hum. Mol. Genet. 1997, 6, 1979–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, J.H.; Yang, L.; Dessaud, E.; Chuang, K.; Moore, D.M.; Rohatgi, R.; Briscoe, J.; Novitch, B.G. Notch Activity Modulates the Responsiveness of Neural Progenitors to Sonic Hedgehog Signaling. Dev. Cell 2015, 33, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishna, U.; Wild, A.; Grzeschik, K.H.; Antonarakis, S.E. Mutation in GLI3 in Postaxial Polydactyly Type A. Nat. Genet. 1997, 17, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishna, U.; Bornholdt, D.; Scott, H.S.; Patel, U.C.; Rossier, C.; Engel, H.; Bottani, A.; Chandal, D.; Blouin, J.L.; Solanki, J.V.; et al. The Phenotypic Spectrum of GLI3 Morphopathies Includes Autosomal Dominant Preaxial Polydactyly Type-IV and Postaxial Polydactyly Type-A/B; No Phenotype Prediction from the Position of GLI3 Mutations. Am. J. Hum. Genet. 1999, 65, 645–655. [Google Scholar] [CrossRef] [PubMed]
- França, M.M.; Jorge, A.A.L.; Carvalho, L.R.S.; Costalonga, E.F.; Vasques, G.A.; Leite, C.C.; Mendonca, B.B.; Arnhold, I.J.P. Novel Heterozygous Nonsense GLI2 Mutations in Patients with Hypopituitarism and Ectopic Posterior Pituitary Lobe without Holoprosencephaly. J. Clin. Endocrinol. Metab. 2010, 95, E384–E391. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Du, Y.-Z.; Mullor, J.L.; Casas, E.; Allen, W.P.; Gillessen-Kaesbach, G.; Roeder, E.R.; Ming, J.E.; I Altaba, A.R.; Muenke, M. Loss-of-Function Mutations in the Human GLI2 Gene Are Associated with Pituitary Anomalies and Holoprosencephaly-like Features. Proc. Natl. Acad. Sci. USA 2003, 100, 13424–13429. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Integrative Genomic Analyses on GLI1: Positive Regulation of GLI1 by Hedgehog-GLI, TGFbeta-Smads, and RTK-PI3K-AKT Signals, and Negative Regulation of GLI1 by Notch-CSL-HES/HEY, and GPCR-Gs-PKA Signals. Int. J. Oncol. 2009, 35, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; I Altaba, A.R. Melanomas Require HEDGEHOG-GLI Signaling Regulated by Interactions between GLI1 and the RAS-MEK/AKT Pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Ohta, M.; Asaoka, Y.; Ikenoue, T.; Tada, M.; Miyabayashi, K.; Mohri, D.; Tanaka, Y.; Ijichi, H.; Tateishi, K.; et al. Regulation of the Hedgehog Signaling by the Mitogen-Activated Protein Kinase Cascade in Gastric Cancer. Mol. Carcinog. 2009, 48, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 Is Regulated through Smoothened-Independent Mechanisms in Neoplastic Pancreatic Ducts and Mediates PDAC Cell Survival and Transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.Y.; Curran, T. The Hedgehog’s Tale: Developing Strategies for Targeting Cancer. Nat. Rev. Cancer 2011, 11, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The Crosstalk of MTOR/S6K1 and Hedgehog Pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Regl, G.; Eichberger, T.; Frischauf, A.-M.; Aberger, F. Efficient Manipulation of Hedgehog/GLI Signaling Using Retroviral Expression Systems. Methods Mol. Biol. 2007, 397, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Bercury, K.K.; Dai, J.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional Ablation of Raptor or Rictor Has Differential Impact on Oligodendrocyte Differentiation and CNS Myelination. J. Neurosci. 2014, 34, 4466–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrun-Julien, F.; Bachmann, L.; Norrmén, C.; Trötzmüller, M.; Köfeler, H.; Rüegg, M.A.; Hall, M.N.; Suter, U. Balanced MTORC1 Activity in Oligodendrocytes Is Required for Accurate CNS Myelination. J. Neurosci. 2014, 34, 8432–8448. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.E.; McLane, L.E.; Bercury, K.K.; Macklin, W.B.; Wood, T.L. Mammalian Target of Rapamycin Promotes Oligodendrocyte Differentiation, Initiation and Extent of CNS Myelination. J. Neurosci. 2014, 34, 4453–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Jiang, W.; Wang, J.; Li, Z.; Zhang, J.; Bu, J.; Zou, J.; Zhou, L.; Yu, S.; Cui, Y.; et al. Oligodendrocyte Precursor Cell-Intrinsic Effect of Rheb1 Controls Differentiation and Mediates MTORC1-Dependent Myelination in Brain. J. Neurosci. 2014, 34, 15764–15778. [Google Scholar] [CrossRef] [PubMed]
- Tyler, W.A.; Gangoli, N.; Gokina, P.; Kim, H.A.; Covey, M.; Levison, S.W.; Wood, T.L. Activation of the Mammalian Target of Rapamycin (MTOR) Is Essential for Oligodendrocyte Differentiation. J. Neurosci. 2009, 29, 6367–6378. [Google Scholar] [CrossRef] [PubMed]
- Aronova, S.; Wedaman, K.; Aronov, P.A.; Fontes, K.; Ramos, K.; Hammock, B.D.; Powers, T. Regulation of Ceramide Biosynthesis by TOR Complex 2. Cell Metab. 2008, 7, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingham, P.W.; Nakano, Y.; Seger, C. Mechanisms and Functions of Hedgehog Signaling across the Metazoa. Nat. Rev. Genet. 2011, 12, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P. Phosphoinositide 3-Kinase and Akt Are Essential for Sonic Hedgehog Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Luo, J.; Mosley, Y.-Y.C.; Hedrick, V.E.; Paul, L.N.; Chang, J.; Zhang, G.; Wang, Y.-K.; Banko, M.R.; Brunet, A.; et al. AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep. 2015, 12, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Drannik, A.; Jiang, F.; Peterson, R.; Turnbull, J. Crosstalk between Notch and Sonic Hedgehog Signaling in a Mouse Model of Amyotrophic Lateral Sclerosis. Neuroreport 2017, 28, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Juryńczyk, M.; Jurewicz, A.; Bielecki, B.; Raine, C.S.; Selmaj, K. Inhibition of Notch Signaling Enhances Tissue Repair in an Animal Model of Multiple Sclerosis. J. Neuroimmunol. 2005, 170, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Juryńczyk, M.; Selmaj, K. Notch: A New Player in MS Mechanisms. J. Neuroimmunol. 2010, 218, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Saher, G.; Brügger, B.; Lappe-Siefke, C.; Möbius, W.; Tozawa, R.; Wehr, M.C.; Wieland, F.; Ishibashi, S.; Nave, K.-A. High Cholesterol Level Is Essential for Myelin Membrane Growth. Nat. Neurosci. 2005, 8, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Vallett, S.M.; Sanchez, H.B.; Rosenfeld, J.M.; Osborne, T.F. A Direct Role for Sterol Regulatory Element Binding Protein in Activation of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Gene. J. Biol. Chem. 1996, 271, 12247–12253. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.K.; Toth, J.I.; Osborne, T.F. Selective association of sterol regulatory element-binding protein isoforms with target promoters in vivo. J. Biol. Chem. 2004, 279, 37360–37367. [Google Scholar] [CrossRef] [PubMed]
- Tabor, D.E.; Kim, J.B.; Spiegelman, B.M.; Edwards, P.A. Identification of Conserved Cis-Elements and Transcription Factors Required for Sterol-Regulated Transcription of Stearoyl-CoA Desaturase 1 and 2. J. Biol. Chem. 1999, 274, 20603–20610. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Fink, M.; Waterman, M.R.; Rozman, D. A CAMP-Responsive Element Binding Site Is Essential for Sterol Regulation of the Human Lanosterol 14alpha-Demethylase Gene (CYP51). Mol. Endocrinol. 2002, 16, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Yamamoto, J.; Okamura, M.; Fujino, T.; Takahashi, S.; Takeuchi, K.; Osborne, T.T.; Yamamoto, T.T. Transcriptional regulation of the murine acetyl CoA synthetase 1 gene through multiple clustered binding sites for SREBPs and a single neighboring site for Sp1. J Biol. Chem. 2001, 276, 34259–34269. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, A.V.; Sharpe, L.J.; Brown, A.J. The Sterol-Based Transcriptional Control of Human 7-Dehydrocholesterol Reductase (DHCR7): Evidence of a Cooperative Regulatory Program in Cholesterol Synthesis. Biochim. Biophys. Acta 2014, 1842, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; DeBose-Boyd, R.A. Regulation of Cholesterol and Fatty Acid Synthesis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004754. [Google Scholar] [CrossRef] [PubMed]
- Norrmèn, C.; Figlia, G.; Lebrun-Julien, F.; Pereira, J.A.; Trötzmüller, M.; Köfeler, H.C.; Rantanen, V.; Wessig, C.; Van Deijk, A.-L.F.; Smit, A.B.; et al. MTORC1 Controls PNS Myelination along the MTORC1-RxRγ-SREBP-Lipid Biosynthesis Axis in Schwann Cells. Cell Rep. 2014, 9, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Saher, G.; Quintes, S.; Möbius, W.; Wehr, M.C.; Krämer-Albers, E.-M.; Brügger, B.; Nave, K.-A. Cholesterol Regulates the Endoplasmic Reticulum Exit of the Major Membrane Protein P0 Required for Peripheral Myelin Compaction. J. Neurosci. 2009, 29, 6094–6104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by MTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of MTOR Complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, I.; Kornberg, T.B. Hedgehog and Its Circuitous Journey from Producing to Target Cells. Semin. Cell Dev. Biol. 2014, 33, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Schmiege, P.; Coutavas, E.; Wang, J.; Li, X. Structures of Human Patched and Its Complex with Native Palmitoylated Sonic Hedgehog. Nature 2018, 560, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 Disease Gene: Homology to Mediators of Cholesterol Homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidet, M.; Joubert, O.; Lacombe, B.; Ciantar, M.; Nehmé, R.; Mollat, P.; Brétillon, L.; Faure, H.; Bittman, R.; Ruat, M.; et al. The Hedgehog Receptor Patched Is Involved in Cholesterol Transport. PLoS ONE 2011, 6, e23834. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Lieberman, A.P. Npc1 Acting in Neurons and Glia Is Essential for the Formation and Maintenance of CNS Myelin. PLoS Genet. 2013, 9, e1003462. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Gruenberg, J.; Roelink, H. Sonic Hedgehog Induces the Segregation of Patched and Smoothened in Endosomes. Curr. Biol. 2002, 12, 983–995. [Google Scholar] [CrossRef] [Green Version]
- Khaliullina, H.; Panáková, D.; Eugster, C.; Riedel, F.; Carvalho, M.; Eaton, S. Patched Regulates Smoothened Trafficking Using Lipoprotein-Derived Lipids. Development 2009, 136, 4111–4121. [Google Scholar] [CrossRef] [PubMed]
- Blassberg, R.; Macrae, J.I.; Briscoe, J.; Jacob, J. Reduced Cholesterol Levels Impair Smoothened Activation in Smith-Lemli-Opitz Syndrome. Hum. Mol. Genet. 2016, 25, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.F.X.; Sircar, R.; Miller, P.S.; Hedger, G.; Luchetti, G.; Nachtergaele, S.; Tully, M.D.; Mydock-McGrane, L.; Covey, D.F.; Rambo, R.P.; et al. Structural Basis of Smoothened Regulation by Its Extracellular Domains. Nature 2016, 535, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Furtado, L.V.; Kelley, R.I.; Opitz, J.M. Disorders of Sterol Biosynthesis. Transl. Sci. Rare Dis. 2016, 1, 145–182. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Giovane, A.; Ragnini-Wilson, A. Targeting Smoothened as a New Frontier in the Functional Recovery of Central Nervous System Demyelinating Pathologies. Int. J. Mol. Sci. 2018, 19, 3677. https://doi.org/10.3390/ijms19113677
Del Giovane A, Ragnini-Wilson A. Targeting Smoothened as a New Frontier in the Functional Recovery of Central Nervous System Demyelinating Pathologies. International Journal of Molecular Sciences. 2018; 19(11):3677. https://doi.org/10.3390/ijms19113677
Chicago/Turabian StyleDel Giovane, Alice, and Antonella Ragnini-Wilson. 2018. "Targeting Smoothened as a New Frontier in the Functional Recovery of Central Nervous System Demyelinating Pathologies" International Journal of Molecular Sciences 19, no. 11: 3677. https://doi.org/10.3390/ijms19113677
APA StyleDel Giovane, A., & Ragnini-Wilson, A. (2018). Targeting Smoothened as a New Frontier in the Functional Recovery of Central Nervous System Demyelinating Pathologies. International Journal of Molecular Sciences, 19(11), 3677. https://doi.org/10.3390/ijms19113677