Chronic Exposure to Palmitate Impairs Insulin Signaling in an Intestinal L-cell Line: A Possible Shift from GLP-1 to Glucagon Production



 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
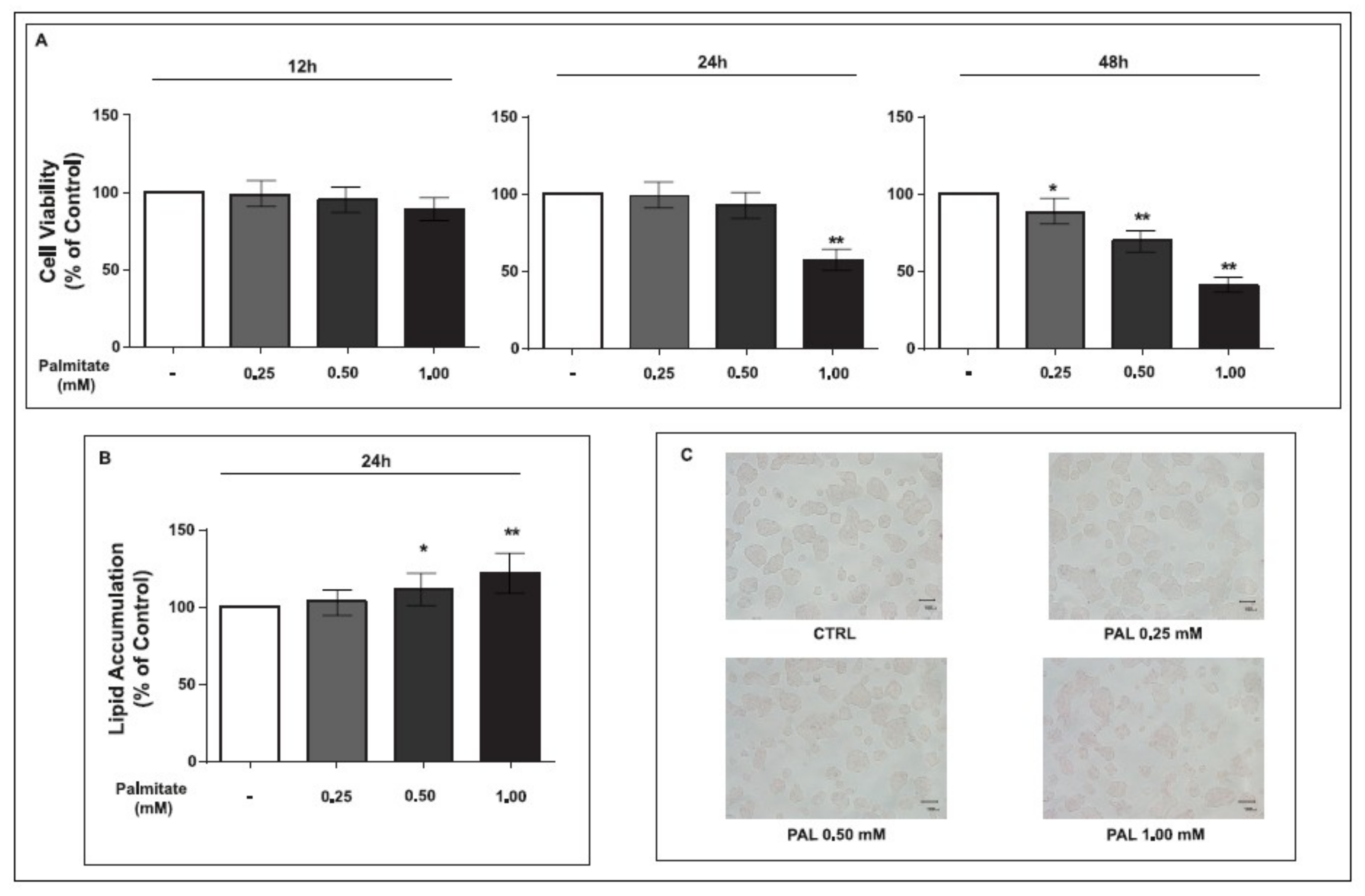
2.1. Cell Viability and Lipid Accumulation of GLUTag Cells Treated with Palmitate
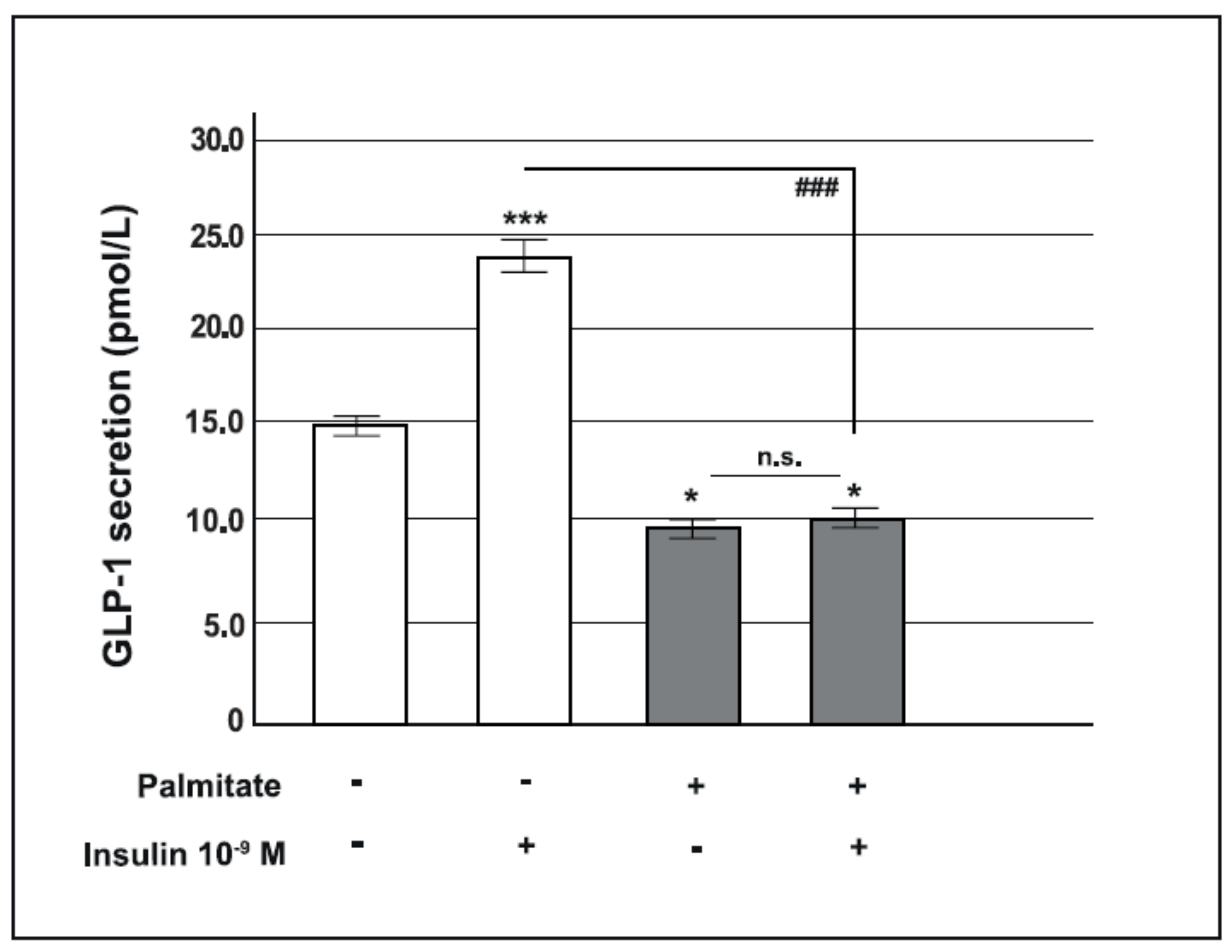
2.2. Chronic Palmitate Exposure Reduced Insulin-Induced GLP-1 Secretion
2.3. Palmitate Impaired IR Phosphorylation and the IRS-1/AKT Pathway
2.4. Palmitate Increased the MAPK Pathway
2.5. Palmitate Up-regulated the IR-A/IR-B Ratio
2.6. Palmitate Increased PAX6 and Proglucagon Expression
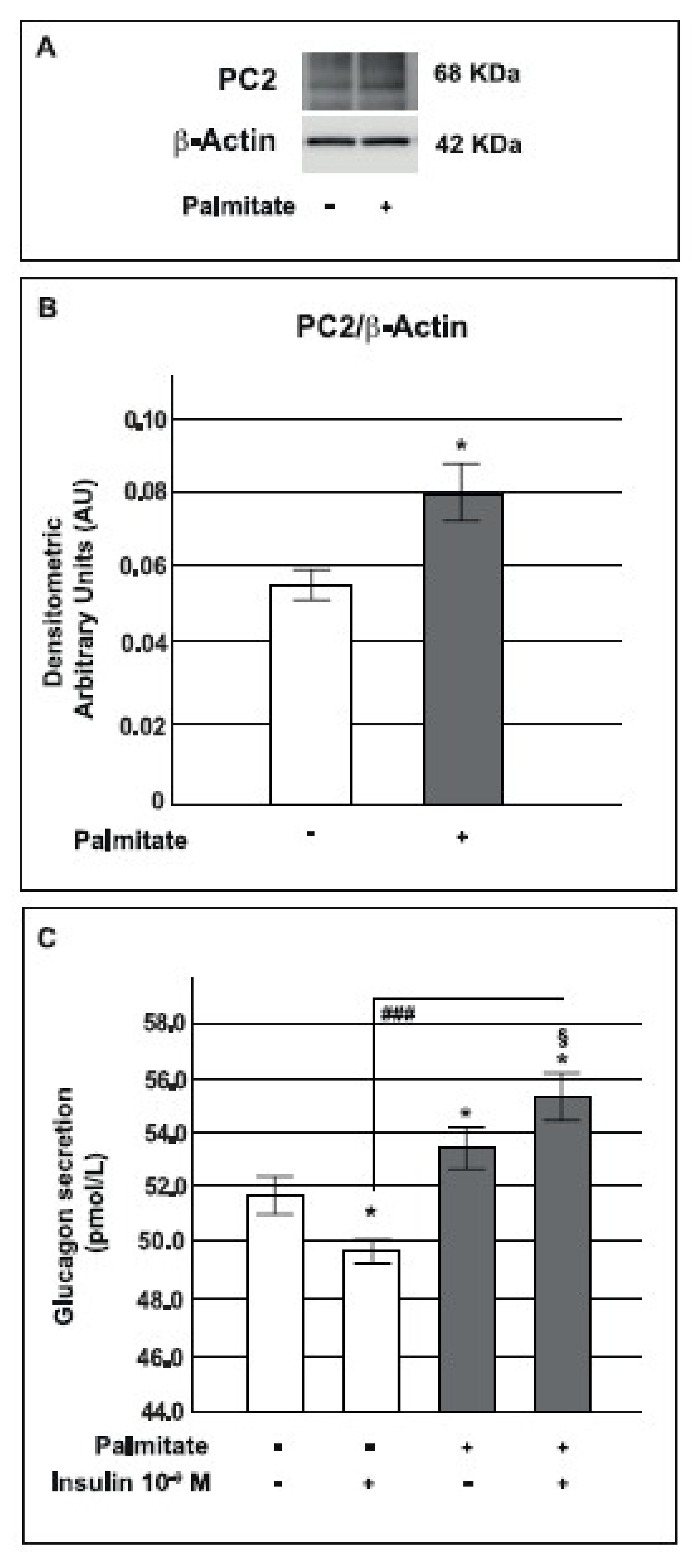
2.7. Palmitate Increased Prohormone Convertase PC2 Expression and Glucagon Secretion
2.8. Palmitate Up-Regulated the MAPK Pathway and Its Downstream Targets
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chronic Exposure to Palmitate
4.3. MTT Cell Viability Assay
4.4. Evaluation of Lipid Accumulation by Nile Red and Oil Red O Staining
4.5. Insulin Stimulation
4.6. GLP-1 and Glucagon Secretion
4.7. Cell Lysis, Immunoprecipitation and Western Blot Analysis
4.8. Total RNA Isolation, Reverse Transcription and Real-time PCR
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Meier, J.J.; Nauck, M.A. Glucagon-like peptide 1(GLP-1) in biology and pathology. Diabetes/Metab. Res. Rev. 2005, 21, 91–117. [Google Scholar] [CrossRef] [PubMed]
- Elrick, H.; Stimmler, L.; Hlad, C.J., Jr.; Arai, Y. Plasma Insulin Response to Oral and Intravenous Glucose Administration. J. Clin. Endocrinol. Metab. 1964, 24, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Skow, M.A.; Bergmann, N.C.; Knop, F.K. Diabetes and obesity treatment based on dual incretin receptor activation: ‘Twincretins’. Diabetes Obes. Metab. 2016, 18, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Gribble, F.M.; Reimann, F. Stimulation of incretin secreting cells. Ther. Adv. Endocrinol. Metab. 2016, 7, 24–42. [Google Scholar] [CrossRef] [PubMed]
- Albrechtsen, N.J.W.; Kuhre, R.E.; Deacon, C.F.; Holst, J.J. Targeting the intestinal L-cell for obesity and type 2 diabetes treatment. Exp. Rev. Endocrinol. Metab. 2014, 9, 61–72. [Google Scholar] [CrossRef]
- Lim, G.E.; Huang, G.J.; Flora, N.; LeRoith, D.; Rhodes, C.J.; Brubaker, P.L. Insulin regulates glucagon-like peptide-1 secretion from the enteroendocrine L cell. Endocrinology 2009, 150, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.; Sauve, M.; Zhao, W.; Stacey, H.M.; Wiber, S.C.; Bolz, S.S.; Brubaker, P.L. Chronic Exposure to TNFalpha Impairs Secretion of Glucagon-Like Peptide-1. Endocrinology 2015, 156, 3950–3960. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.; Patrone, C.; Holst, J.J.; Zhang, Q.; Sjoholm, A. Metformin protects against lipoapoptosis and enhances GLP-1 secretion from GLP-1-producing cells. J. Gastroenterol. 2013, 48, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Pietraszek, A.; Gregersen, S.; Pedersen, S.B.; Holst, J.J.; Hermansen, K. Acute effects of monounsaturated fat on postprandial lipemia and gene expression in first-degree relatives of subjects with type 2 diabetes. Eur. J. Clin. Nutr. 2014, 68, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, C.; Rasmussen, O.; Lousen, T.; Holst, J.J.; Fenselau, S.; Schrezenmeir, J.; Hermansen, K. Differential effects of saturated and monounsaturated fatty acids on postprandial lipemia and incretin responses in healthy subjects. Am. J. Clin. Nutr. 1999, 69, 1135–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimann, F. Molecular mechanisms underlying nutrient detection by incretin-secreting cells. Int. Dairy J. 2010, 20, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Tolhurst, G.; Reimann, F.; Gribble, F.M. Nutritional regulation of glucagon-like peptide-1 secretion. J. Physiol. 2009, 587, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbano, F.; Filippello, A.; Di Pino, A.; Barbagallo, D.; Di Mauro, S.; Pappalardo, A.; Rabuazzo, A.M.; Purrello, M.; Purrello, F.; Piro, S. Altered expression of uncoupling protein 2 in GLP-1-producing cells after chronic high glucose exposure: Implications for the pathogenesis of diabetes mellitus. Am. J. Physiol. Cell Physiol. 2016, 310, C558–C567. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.J.; Habener, J.F. The glucagon-like peptides. Endocr. Rev. 1999, 20, 876–913. [Google Scholar] [CrossRef] [PubMed]
- Eissele, R.; Goke, R.; Willemer, S.; Harthus, H.P.; Vermeer, H.; Arnold, R.; Goke, B. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur. J. Clin. Investig. 1992, 22, 283–291. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Hamaguchi, K.; Seike, M.; Himeno, K.; Sakata, T.; Yoshimatsu, H. Role of fatty acid composition in the development of metabolic disorders in sucrose-induced obese rats. Exp. Biol. Med. 2004, 229, 486–493. [Google Scholar] [CrossRef]
- Vasu, S.; Moffett, R.C.; McClenaghan, N.H.; Flatt, P.R. Differential molecular and cellular responses of GLP-1 secreting L-cells and pancreatic alpha cells to glucotoxicity and lipotoxicity. Exp. Cell Res. 2015, 336, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.; Pais, R.; Habib, A.M.; Brighton, C.A.; Yeo, G.S.; Reimann, F.; Gribble, F.M. High fat diet impairs the function of glucagon-like peptide-1 producing L-cells. Peptides 2016, 77, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappe, C.; Holst, J.J.; Zhang, Q.; Sjoholm, A. Molecular mechanisms of lipoapoptosis and metformin protection in GLP-1 secreting cells. Biochem. Biophys. Res. Commun. 2012, 427, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambo, V.; Simon-Szabo, L.; Szelenyi, P.; Kereszturi, E.; Banhegyi, G.; Csala, M. Lipotoxicity in the liver. World J. Hepatol. 2013, 5, 550–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Krssak, M.; Falk Petersen, K.; Dresner, A.; DiPietro, L.; Vogel, S.M.; Rothman, D.L.; Roden, M.; Shulman, G.I. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1H NMR spectroscopy study. Diabetologia 1999, 42, 113–116. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr. Diabetes Rep. 2010, 10, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piro, S.; Maniscalchi, E.T.; Monello, A.; Pandini, G.; Mascali, L.G.; Rabuazzo, A.M.; Purrello, F. Palmitate affects insulin receptor phosphorylation and intracellular insulin signal in a pancreatic alpha-cell line. Endocrinology 2010, 151, 4197–4206. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Song, J.; Liu, H.; Yan, F.; He, T.; Wang, L.; Shen, H.; Hou, X.; Chen, L. Insulin regulates glucagon-like peptide-1 secretion by pancreatic alpha cells. Endocrine 2018, 62, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Rhee, N.A.; Wahlgren, C.D.; Pedersen, J.; Mortensen, B.; Langholz, E.; Wandall, E.P.; Friis, S.U.; Vilmann, P.; Paulsen, S.J.; Kristiansen, V.B.; et al. Effect of Roux-en-Y gastric bypass on the distribution and hormone expression of small-intestinal enteroendocrine cells in obese patients with type 2 diabetes. Diabetologia 2015, 58, 2254–2258. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Bagger, J.I.; Wewer Albrechtsen, N.J.; Christensen, M.; Grondahl, M.; Hartmann, B.; Mathiesen, E.R.; Hansen, C.P.; Storkholm, J.H.; van Hall, G.; et al. Evidence of Extrapancreatic Glucagon Secretion in Man. Diabetes 2016, 65, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Vranic, M.; Pek, S.; Kawamori, R. Increased “glucagon immunoreactivity” in plasma of totally depancreatized dogs. Diabetes 1974, 23, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Knop, F.K. Extrapancreatic glucagon: Present status. Diabetes Res. Clin. Pract. 2018. [Google Scholar] [CrossRef] [PubMed]
- Karesen, R.; Tronier, B.; Aune, S. Immunoreactive glucagon and insulin C-peptide in man after resection of the pancreas and total pancreatectomy. Am. J. Surg. 1980, 140, 272–276. [Google Scholar] [CrossRef]
- Holst, J.J.; Pedersen, J.H.; Baldissera, F.; Stadil, F. Circulating glucagon after total pancreatectomy in man. Diabetologia 1983, 25, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Zhou, A.; Webb, G.; Carroll, R.; Ravazzola, M.; Orci, L.; Steiner, D.F. Severe defect in proglucagon processing in islet A-cells of prohormone convertase 2 null mice. J. Biol. Chem. 2001, 276, 27197–27202. [Google Scholar] [CrossRef] [PubMed]
- Knop, F.K. Resolution of type 2 diabetes following gastric bypass surgery: Involvement of gut-derived glucagon and glucagonotropic signalling? Diabetologia 2009, 52, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Yang, H.S.; Yu, T.; Shan, T.D.; Li, J.Y.; Huang, C.Z.; Zhong, W.; Xia, Z.S.; Chen, Q.K. MEK/ERK pathway activation by insulin receptor isoform alteration is associated with the abnormal proliferation and differentiation of intestinal epithelial cells in diabetic mice. Mol. Cell. Biochem. 2016, 413, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, S.E.; Giel-Moloney, M.; Leiter, A.B. Minireview: Development and differentiation of gut endocrine cells. Endocrinology 2004, 145, 2639–2644. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, I.; Bruun, J.A.; Bjorkoy, G.; Holm, T.; Johansen, T. Phosphorylation of the transactivation domain of Pax6 by extracellular signal-regulated kinase and p38 mitogen-activated protein kinase. J. Biol. Chem. 1999, 274, 15115–15126. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.E.; Asa, S.L.; Drucker, D.J. Essential requirement for Pax6 in control of enteroendocrine proglucagon gene transcription. Mol. Endocrinol. 1999, 13, 1474–1486. [Google Scholar] [CrossRef] [PubMed]
- Rouille, Y.; Kantengwa, S.; Irminger, J.C.; Halban, P.A. Role of the prohormone convertase PC3 in the processing of proglucagon to glucagon-like peptide 1. J. Biol. Chem. 1997, 272, 32810–32816. [Google Scholar] [CrossRef] [PubMed]
- Wideman, R.D.; Covey, S.D.; Webb, G.C.; Drucker, D.J.; Kieffer, T.J. A switch from prohormone convertase (PC)-2 to PC1/3 expression in transplanted alpha-cells is accompanied by differential processing of proglucagon and improved glucose homeostasis in mice. Diabetes 2007, 56, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H. Pancreas: Extrapancreatic glucagon in humans. Nat. Rev. Endocrinol. 2016, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Lund, A. On the role of the gut in diabetic hyperglucagonaemia. Dan. Med. J. 2017, 64, 5340. [Google Scholar]
- Kuhre, R.E.; Wewer Albrechtsen, N.J.; Deacon, C.F.; Balk-Moller, E.; Rehfeld, J.F.; Reimann, F.; Gribble, F.M.; Holst, J.J. Peptide production and secretion in GLUTag, NCI-H716, and STC-1 cells: A comparison to native L-cells. J. Mol. Endocrinol. 2016, 56, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Jorsal, T.; Rhee, N.A.; Pedersen, J.; Wahlgren, C.D.; Mortensen, B.; Jepsen, S.L.; Jelsing, J.; Dalboge, L.S.; Vilmann, P.; Hassan, H.; et al. Enteroendocrine K and L cells in healthy and type 2 diabetic individuals. Diabetologia 2018, 61, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Damholt, A.B.; Buchan, A.M.; Holst, J.J.; Kofod, H. Proglucagon processing profile in canine L cells expressing endogenous prohormone convertase 1/3 and prohormone convertase 2. Endocrinology 1999, 140, 4800–4808. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.S.; Gosmain, Y.; Marthinet, E.; Philippe, J. Pax6 regulates the proglucagon processing enzyme PC2 and its chaperone 7B2. Mol. Cell. Biol. 2009, 29, 2322–2334. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Asa, S.L.; Drucker, D.J. Glucagon gene 5’-flanking sequences direct expression of simian virus 40 large T antigen to the intestine, producing carcinoma of the large bowel in transgenic mice. J. Biol. Chem. 1992, 267, 10705–10708. [Google Scholar] [PubMed]
- Brubaker, P.L.; Schloos, J.; Drucker, D.J. Regulation of glucagon-like peptide-1 synthesis and secretion in the GLUTag enteroendocrine cell line. Endocrinology 1998, 139, 4108–4114. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.; Zhang, Q.; Holst, J.J.; Nystrom, T.; Sjoholm, A. Evidence for paracrine/autocrine regulation of GLP-1-producing cells. American journal of physiology. Cell Physiol. 2013, 305, C1041–C1049. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Jin, T.; Asa, S.L.; Young, T.A.; Brubaker, P.L. Activation of proglucagon gene transcription by protein kinase-A in a novel mouse enteroendocrine cell line. Mol. Endocrinol. 1994, 8, 1646–1655. [Google Scholar] [PubMed]
- Boren, J.; Taskinen, M.R.; Olofsson, S.O.; Levin, M. Ectopic lipid storage and insulin resistance: A harmful relationship. J. Intern. Med. 2013, 274, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Era, S.; Bhamidipati, S.P.; Reed, R.G. Locations of the three primary binding sites for long-chain fatty acids on bovine serum albumin. Proc. Natl. Acad. Sci. USA 1991, 88, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Bassingthwaighte, J.B.; Noodleman, L.; van der Vusse, G.; Glatz, J.F. Modeling of palmitate transport in the heart. Mol. Cell. Biochem. 1989, 88, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed]
- Bobo, R.C.; Partin, J.C.; Schubert, W.K.; Saalfeld, K. Abnormal lipid accumulation within the small intestinal mucosa of children with juvenile-onset diabetes mellitus. Am. J. Dis. Child. 1977, 131, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Douglass, J.D.; Malik, N.; Chon, S.H.; Wells, K.; Zhou, Y.X.; Choi, A.S.; Joseph, L.B.; Storch, J. Intestinal mucosal triacylglycerol accumulation secondary to decreased lipid secretion in obese and high fat fed mice. Front. Physiol. 2012, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.L.; Zhang, Y.; Liu, J.; Wu, Y.; Hu, Z.B.; Liu, L.; Liu, B.C. Inflammatory stress induces lipid accumulation in multi-organs of db/db mice. Acta Biochim. Biophys. Sin. 2015, 47, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Yamada, R.; Das, S.S.; Sato, T.; Takahashi, A.; Hiratsuka, M.; Hirasawa, N. Glucagon-like peptide-1 production in the GLUTag cell line is impaired by free fatty acids via endoplasmic reticulum stress. Metab. Clin. Exp. 2014, 63, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xiao, K.L.; Chen, L.; Wu, C.; Hu, X.; Zeng, T.; Chen, X.Q.; Li, W.J.; Deng, X.; Li, H.; et al. Insulin sensitizers improve the GLP-1 secretion and the amount of intestinal L cells on high-fat-diet-induced catch-up growth. Nutrition 2017, 39–40, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Haering, M.F.; Fujisaka, S.; Lutter, D.; Lee, K.Y.; Li, N.; Gerber, G.K.; Bry, L.; Kahn, C.R. Regulation of Glucose Uptake and Enteroendocrine Function by the Intestinal Epithelial Insulin Receptor. Diabetes 2017, 66, 886–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.e6. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Chiang, J.Y.; Ding, W.X.; Dunn, W.; Roberts, B.; Li, T. Saturated fatty acids activate ERK signaling to downregulate hepatic sortilin 1 in obese and diabetic mice. J. Lipid Res. 2013, 54, 2754–2762. [Google Scholar] [CrossRef] [PubMed]
- Swisa, A.; Glaser, B.; Dor, Y. Metabolic Stress and Compromised Identity of Pancreatic Beta Cells. Front. Genet. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Shaham, O.; Menuchin, Y.; Farhy, C.; Ashery-Padan, R. Pax6: A multi-level regulator of ocular development. Prog. Retin. Eye Res. 2012, 31, 351–376. [Google Scholar] [CrossRef] [PubMed]
- Osumi, N.; Shinohara, H.; Numayama-Tsuruta, K.; Maekawa, M. Concise review: Pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem Cells 2008, 26, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Gosmain, Y.; Cheyssac, C.; Heddad Masson, M.; Dibner, C.; Philippe, J. Glucagon gene expression in the endocrine pancreas: The role of the transcription factor Pax6 in alpha-cell differentiation, glucagon biosynthesis and secretion. Diabetes Obes. Metab. 2011, 13 (Suppl. 1), 31–38. [Google Scholar] [CrossRef] [PubMed]
- Gil-Lozano, M.; Wu, W.K.; Martchenko, A.; Brubaker, P.L. High-Fat Diet and Palmitate Alter the Rhythmic Secretion of Glucagon-Like Peptide-1 by the Rodent L-cell. Endocrinology 2016, 157, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhou, A.; Dey, A.; Norrbom, C.; Carroll, R.; Zhang, C.; Laurent, V.; Lindberg, I.; Ugleholdt, R.; Holst, J.J.; et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc. Natl. Acad. Sci. USA 2002, 99, 10293–10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, P.K.; Goodman, R.H.; Dee, P.C.; Habener, J.F. Pancreatic preproglucagon cDNA contains two glucagon-related coding sequences arranged in tandem. Proc. Natl. Acad. Sci. USA 1982, 79, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Rouille, Y.; Martin, S.; Steiner, D.F. Differential processing of proglucagon by the subtilisin-like prohormone convertases PC2 and PC3 to generate either glucagon or glucagon-like peptide. J. Biol. Chem. 1995, 270, 26488–26496. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Burcelin, R. GLP-1 effects on islets: Hormonal, neuronal, or paracrine? Diabetes Care 2013, 36 (Suppl. 2), S145–S148. [Google Scholar] [CrossRef] [PubMed]
- Schuh, K.; Pahl, A. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochem. Pharmacol. 2009, 77, 1827–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, J.H.; Jetten, A.M. NF-kappaB-dependent transcriptional activation in lung carcinoma cells by farnesol involves p65/RelA(Ser276) phosphorylation via the MEK-MSK1 signaling pathway. J. Biol. Chem. 2008, 283, 16391–16399. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Lee, Y.C.; Asa, S.L.; Brubaker, P.L. Inhibition of pancreatic glucagon gene expression in mice bearing a subcutaneous glucagon-producing GLUTag transplantable tumor. Mol. Endocrinol. 1992, 6, 2175–2184. [Google Scholar] [PubMed]
- Di Mauro, S.; Ragusa, M.; Urbano, F.; Filippello, A.; Di Pino, A.; Scamporrino, A.; Pulvirenti, A.; Ferro, A.; Rabuazzo, A.M.; Purrello, M.; et al. Intracellular and extracellular miRNome deregulation in cellular models of NAFLD or NASH: Clinical implications. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile red: A selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Yi, J.; Liang, F.; Jiang, B.; Xiao, Y.; Gao, S.; Yang, N.; Hu, H.; Xie, W.F.; Chen, W. X-3, a mangiferin derivative, stimulates AMP-activated protein kinase and reduces hyperglycemia and obesity in db/db mice. Mol. Cell. Endocrinol. 2015, 405, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Koppensteiner, P.; Trinchese, F.; Fa, M.; Puzzo, D.; Gulisano, W.; Yan, S.; Poussin, A.; Liu, S.; Orozco, I.; Dale, E.; et al. Time-dependent reversal of synaptic plasticity induced by physiological concentrations of oligomeric Abeta42: An early index of Alzheimer’s disease. Sci. Rep. 2016, 6, 32553. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Denton, R.M. Insulin stimulation of pyruvate dehydrogenase in adipocytes involves two distinct signalling pathways. Biochem. J. 2003, 369 Pt 2, 351–356. [Google Scholar] [CrossRef]
- Urbano, F.; Bugliani, M.; Filippello, A.; Scamporrino, A.; Di Mauro, S.; Di Pino, A.; Scicali, R.; Noto, D.; Rabuazzo, A.M.; Averna, M.; et al. Atorvastatin but Not Pravastatin Impairs Mitochondrial Function in Human Pancreatic Islets and Rat β-Cells. Direct Effect of Oxidative Stress. Sci. Rep. 2017, 7, 11863. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippello, A.; Urbano, F.; Di Mauro, S.; Scamporrino, A.; Di Pino, A.; Scicali, R.; Rabuazzo, A.M.; Purrello, F.; Piro, S. Chronic Exposure to Palmitate Impairs Insulin Signaling in an Intestinal L-cell Line: A Possible Shift from GLP-1 to Glucagon Production. Int. J. Mol. Sci. 2018, 19, 3791. https://doi.org/10.3390/ijms19123791
Filippello A, Urbano F, Di Mauro S, Scamporrino A, Di Pino A, Scicali R, Rabuazzo AM, Purrello F, Piro S. Chronic Exposure to Palmitate Impairs Insulin Signaling in an Intestinal L-cell Line: A Possible Shift from GLP-1 to Glucagon Production. International Journal of Molecular Sciences. 2018; 19(12):3791. https://doi.org/10.3390/ijms19123791
Chicago/Turabian StyleFilippello, Agnese, Francesca Urbano, Stefania Di Mauro, Alessandra Scamporrino, Antonino Di Pino, Roberto Scicali, Agata Maria Rabuazzo, Francesco Purrello, and Salvatore Piro. 2018. "Chronic Exposure to Palmitate Impairs Insulin Signaling in an Intestinal L-cell Line: A Possible Shift from GLP-1 to Glucagon Production" International Journal of Molecular Sciences 19, no. 12: 3791. https://doi.org/10.3390/ijms19123791
APA StyleFilippello, A., Urbano, F., Di Mauro, S., Scamporrino, A., Di Pino, A., Scicali, R., Rabuazzo, A. M., Purrello, F., & Piro, S. (2018). Chronic Exposure to Palmitate Impairs Insulin Signaling in an Intestinal L-cell Line: A Possible Shift from GLP-1 to Glucagon Production. International Journal of Molecular Sciences, 19(12), 3791. https://doi.org/10.3390/ijms19123791