Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities


{kind=link}
{kind=link}
Abstract
:1. Introduction
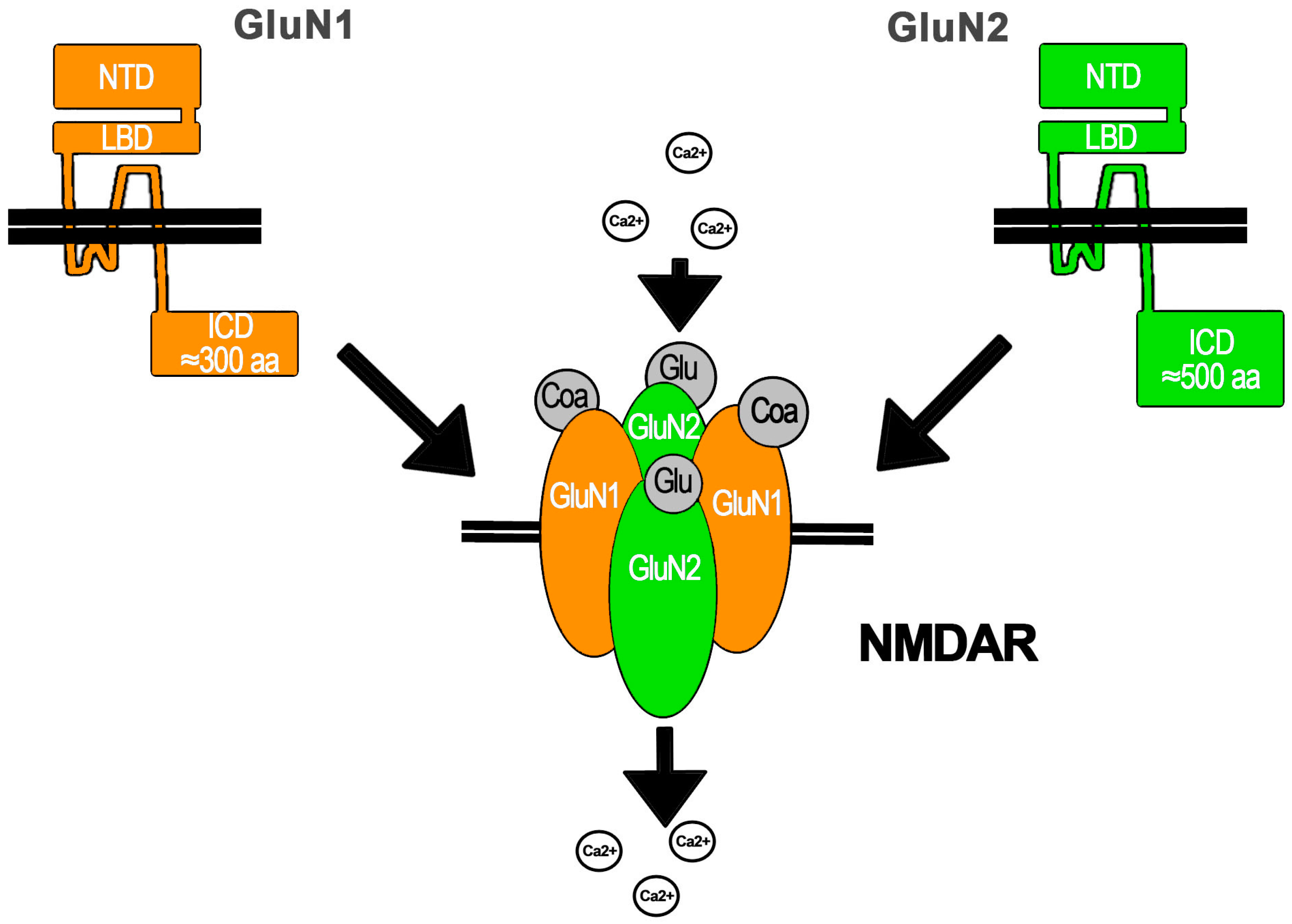
NMDAR Essentials
2. Approaches to Study f-iNMDARs
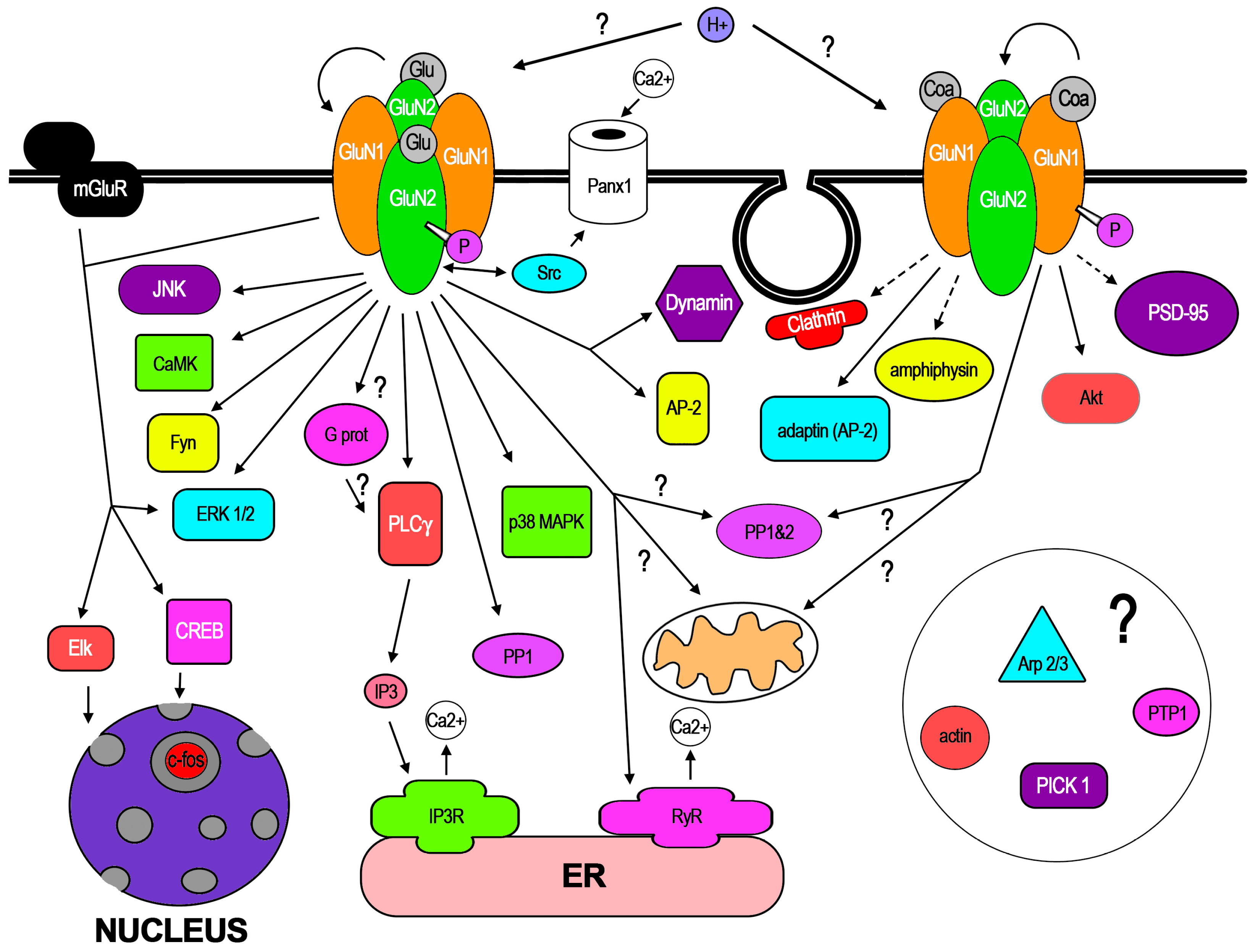
3. Cellular Mechanisms Involved in f-iNMDARs and Its Cellular Effects
3.1. Conformational Changes of NMDAR Subunits
3.2. Ca2+ Dynamics and LTD
3.3. Endocytosis, Signaling Pathways, and Membrane Dynamics
3.4. Neuronal Death and Survival
3.5. Pathologies
4. f-iNMDARs in Astrocytes
5. Insights
5.1. Structure–Function
5.2. Signaling
5.3. Pathology and Clinic
5.4. Evolution
6. Conclusions
Acknowledgements
Conflicts of Interest
References
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca Balderas, P.; Gonzalez Hernandez, J.R. NMDA Receptors in Astroglia: Chronology, Controversies and Contradictions from a Complex Molecule. In Astrocyte Physiology and Pathology; Intech Open: Rijeka, Croatia, 2018. [Google Scholar]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ding, Q.; Chen, Z.; Yun, H.; Wang, H. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-d-aspartate receptor function and neuronal excitotoxicity. J. Biol. Chem. 2013, 288, 24151–24159. [Google Scholar] [CrossRef] [PubMed]
- Chung, C. NMDA receptor as a newly identified member of the metabotropic glutamate receptor family: Clinical implications for neurodegenerative diseases. Mol. Cells 2013, 36, 99–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dore, K.; Stein, I.S.; Brock, J.A.; Castillo, P.E.; Zito, K.; Sjöström, P.J. Unconventional NMDA Receptor Signaling. J. Neurosci. 2017, 37, 10800–10807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dore, K.; Aow, J.; Malinow, R. The emergence of NMDA receptor metabotropic function: Insights from imaging. Front. Synaptic Neurosci. 2016, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W.; Gray, J.A.; Zito, K. Non-ionotropic signaling by the NMDA receptor: Controversy and opportunity. F1000Research 2016, 5, 1010. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [PubMed]
- Aarts, M.M.; Tymianski, M. Novel treatment of excitotoxicity: Targeted disruption of intracellular signalling from glutamate receptors. Biochem. Pharmacol. 2003, 66, 877–886. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Cole, R.L.; Meyers, M.A.; Hyman, S.E. Contrasting calcium dependencies of SAPK and ERK activations by glutamate in cultured striatal neurons. J. Neurochem. 1999, 72, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.; Fox, R.; Alfonso, S.; Aow, J.; Malinow, R. GluA1 trafficking and metabotropic NMDA: Addressing results from other laboratories inconsistent with ours. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 369, 20130145. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca Balderas, P.; Aguilera, P. A Metabotropic-Like Flux-Independent NMDA Receptor Regulates Ca2+ Exit from Endoplasmic Reticulum and Mitochondrial Membrane Potential in Cultured Astrocytes. PLoS ONE 2015, 10, e0126314. [Google Scholar] [CrossRef] [PubMed]
- Dore, K.; Aow, J.; Malinow, R. Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc. Natl. Acad. Sci. USA 2015, 112, 14705–14710. [Google Scholar] [CrossRef] [PubMed]
- Aow, J.; Dore, K.; Malinow, R. Conformational signaling required for synaptic plasticity by the NMDA receptor complex. Proc. Natl. Acad. Sci. USA 2015, 112, 14711–14716. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.Y.M.; Manor, D. Confocal microscopic imaging of [Ca2+] in Cultured Hippocampal Neyrons Following Exposure to N-methyl-d-aspartate. J. Physiol. 1992, 448, 655–676. [Google Scholar] [CrossRef] [PubMed]
- Alford, S.; Frenguellit, B.G.; Schofieldt, J.G.; Collingridge, G.L. Chracterization of Ca2+ Signals Induced in Hippocampal CA1 Neurones by the Synaptic Activation of NMDAR Receptors. J. Physiol. 1993, 469, 693–716. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Colome, A.M.; Ortega, A.; Romo-de-Vivar, M. Excitatory amino acid-induced phosphoinositide hydrolysis in Muller glia. Glia 1993, 9, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Mayford, M.; Wang, J.; Kandel, E.R.; O’Dell, T.J. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell 1995, 81, 891–904. [Google Scholar] [CrossRef]
- Miller, L.D.P.O.; Petrozzino, J.J.; Connor, A. Ca2+ Release From Intracellular Stores Induced by Afferent Stimulation of CA3 Pyramidal Neurons in Hippocmpal Slices. J. Neurophysiol. 1996, 76, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Scanzini, M.; Malenka, R.C.; Nicoll, R.A. Role of Intracellular Interactions in Heterosynaptic Long-term Depression. Nature 1996, 380, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Emptage, N.; Bliss, T.V.P.; Fine, A. Single Synaptic Events Evoke NMDA Receptor—Mediated Release of Calcium from Internal Stores in Hippocampal Dendritic Spines. Neuron 1999, 22, 115–124. [Google Scholar] [CrossRef]
- Nabavi, S.; Kessels, H.W.; Alfonso, S.; Aow, J.; Fox, R.; Malinow, R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4027–4032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Hsu, C.; Cembrowski, M.S.; Mensh, B.D.; Spruston, N. Dendritic sodium spikes are required for long-term potentiation at distal synapses on hippocampal pyramidal neurons. Elife 2015, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Stein, I.S.; Gray, J.A.; Zito, K. Non-Ionotropic NMDA Receptor Signaling Drives Activity-Induced Dendritic Spine Shrinkage. J. Neurosci. 2015, 35, 12303–12308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.C.; Jahr, C.E. Postsynaptic, not presynaptic NMDA receptors are required for spike-timing-dependent LTD induction. Nat. Neurosci. 2016, 19, 1218–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latif-Hernandez, A.; Faldini, E.; Ahmed, T.; Balschun, D. Separate Ionotropic and Metabotropic Glutamate Receptor Functions in Depotentiation vs. LTP: A Distinct Role for Group1 mGluR Subtypes and NMDARs. Front. Cell. Neurosci. 2016, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, T.; Chou, C.Y.C.; Li, S.Y.; Mancino, A.; Costa, R.P.; Brock, J.A.; Nuro, E.; Buchanan, K.A.; Elgar, D.; Blackman, A.V.; et al. Differential Regulation of Evoked and Spontaneous Release by Presynaptic NMDA Receptors. Neuron 2017, 96, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Vissel, B.; Krupp, J.J.; Heinemann, S.F.; Westbrook, G.L. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat. Neurosci. 2001, 4, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Barria, A.; Malinow, R. Subunit-specific NMDA receptor trafficking to synapses. Neuron 2002, 35, 345–353. [Google Scholar] [CrossRef]
- Nong, Y.; Huang, Y.Q.; Ju, W.; Kalia, L.V.; Ahmadian, G.; Wang, Y.T.; Salter, M.W. Glycine binding primes NMDA receptor internalization. Nature 2003, 422, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mao, L.; Tang, Q.; Samdani, S.; Liu, Z.; Wang, J.Q. A novel Ca2+-independent signaling pathway to extracellular signal-regulated protein kinase by coactivation of NMDA receptors and metabotropic glutamate receptor 5 in neurons. J. Neurosci. 2004, 24, 10846–10857. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, R.; Lujan, B.; Chen, J.; Zhang, J. Glycine Potentiates AMPA Receptor Function through Metabotropic Activation of GluN2A-Containing NMDA Receptors. Front. Mol. Sci. 2016, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.S.; Papouin, T.; Ladepeche, L.; Yao, A.; Langlais, V.C.; Bouchet, D.; Dulong, J.; Mothet, J.; Sacchi, S.; Pollegioni, L.; et al. Co-agonists differentially tune GluN2B-NMDA receptor trafficking at hippocampal synapses. Elife 2017, 1, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Murota, S. NMDA receptor stimulation in the absence of extracellular Ca2+ potentiates Ca2+ influx-dependent cell death system. Brain Res. 2005, 1035, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Li, P.; Betts, K.; Liu, R. Two-Photon Imaging of Stroke Onset In Vivo Reveals That NMDA-Receptor Independent Ischemic Depolarization Is the Major Cause of Rapid Reversible Damage to Dendrites and Spines. J. Neurosci. 2008, 28, 1756–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Chen, J.; Lujan, B.; Lei, R.; Zhang, M.; Wang, Z.; Liao, M.; Li, Z.; Wan, Y.; Liu, F.; et al. Glycine triggers a non-ionotropic activity of GluN2A-containing NMDA receptors to confer neuroprotection. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Panatier, A.; Theodosis, D.T.; Mothet, J.P.; Touquet, B.; Pollegioni, L.; Poulain, D.A.; Oliet, S.H.R. Glia-Derived d-Serine Controls NMDA Receptor Activity and Synaptic Memory. Cell 2006, 125, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-term potentiation depends on release of d-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hu, R.; Liao, H.; Zhang, Y.; Lei, R.; Zhang, Z. A non-ionotropic activity of NMDA receptors contributes to glycine- induced neuroprotection in cerebral ischemia-reperfusion injury. Sci. Rep. 2017, 7, 3575. [Google Scholar] [CrossRef] [PubMed]
- Weilinger, N.L.; Lohman, A.W.; Rakai, B.D.; Ma, E.M.M.; Bialecki, J.; Maslieieva, V.; Rilea, T.; Bandet, M.V.; Ikuta, N.T.; Scott, L.; et al. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat. Neurosci. 2016, 19, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Tamburri, A.; Dudilot, A.; Licea, S.; Bourgeois, C.; Boehm, J. NMDA-receptor activation but not ion flux is required for amyloid-beta induced synaptic depression. PLoS ONE 2013, 8, e65350. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.W.; Nabavi, S.; Malinow, R. Metabotropic NMDA receptor function is required for beta-amyloid-induced synaptic depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4033–4038. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Pankratov, Y.; Parpura, V.; Verkhratsky, A. Ionotropic receptors in neuronal-astroglial signalling: What is the role of “excitable” molecules in non-excitable cells. BBA Mol. Cell Res. 2010, 1813, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Palygin, O.; Lalo, U.; Pankratov, Y. Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br. J. Pharmacol. 2011, 163, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Schachner, M. Pharmacological properties of gamma-aminobutyric acid-, glutamate-, and aspartate-induced depolarizations in cultured astrocytes. J. Neurosci. 1985, 5, 3295–3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipke, C.G.; Ohlemeyer, C.; Matyash, M.; Nolte, C.; Kettenmann, H.; Kirchhoff, F. Astrocytes of the mouse neocortex express functional N-methyl-d-aspartate receptors. FASEB J. 2001, 15, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, T.; Matsuoka, T.; Nomura, T.; Kondoh, T.; Tamaki, N.; Okada, Y. Store Ca2+ depletion enhances NMDA responses in cultured human astrocytes. Biochem. Biophys. Res. Commun. 1999, 259, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, T.; Nishizaki, T.; Aihara, H.; Tamaki, N. NMDA-responsible, APV-insensitive receptor in cultured human astrocytes. Life Sci. 2001, 68, 1761–1767. [Google Scholar] [CrossRef]
- Gerard, F.; Hansson, E. Inflammatory activation enhances NMDA-triggered Ca2+ signalling and IL-1beta secretion in primary cultures of rat astrocytes. Brain Res. 2012, 1473, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Blasco, D.; Santofimia-Castaño, P.; Gonzalez, A.; Almeida, A.; Bolaños, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5–Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Ting, K.K.; Adams, S.; Brew, B.J.; Chung, R.; Guillemin, G.J. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS ONE 2010, 5, e14123. [Google Scholar] [CrossRef] [PubMed]
- Eccles, J.C.; Mcgeer, P.L. Ionotropic and metabotropic neurotransmission. Trends Neurosci. 1979, 2, 39–40. [Google Scholar] [CrossRef]
- Valbuena, S.; Lerma, J. Non-canonical Signaling, the Hidden Life of Ligand-Gated Ion Channels. Neuron 2016, 92, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Tenorio, M.; Porras-González, C.; Castellano, A.; Del Valle-Rodríguez, A.; López-Barneo, J.; Ureña, J. Metabotropic regulation of RhoA/Rho-associated kinase by l-type Ca2+ Channels: New Mechanism for Depolarization-Evoked Mammalian Arterial Contraction. Circ. Res. 2011, 108, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Cidad, P.; Jiménez-Pérez, L.; García-Arribas, D.; Miguel-Velado, E.; Tajada, S.; Ruiz-Mcdavitt, C.; López-López, J.R.; Pérez-García, M.T. Kv1.3 channels can modulate cell proliferation during phenotypic switch by an ion-flux independent mechanism. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.B.; Koeppe, R.E.; Andersen, O.S. Induction of Conductance Heterogeneity in Gramicidin Channels. Biochemistry 1989, 28, 6571–6583. [Google Scholar] [CrossRef] [PubMed]
- Mothet, J.-P.; Le Bail, M.; Billard, J.-M. Time and space profiling of NMDAR co-agonist functions. J. Neurochem. 2015, 135, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wong, A.H.; Liu, F. Interactions between NMDA and dopamine receptors: A potential therapeutic target. Brain Res. 2012, 1476, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.D.; Lester, R.A.; Tong, G.; Jahr, C.E.; Westbrook, G.L. The time course of glutamate in the synaptic cleft. Science 1992, 258, 1498–1501. [Google Scholar] [CrossRef] [PubMed]
- Meisenbock, G.; De Angelis, D.A.; Rothman, J.E. Visualizing sectretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 1998, 394, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, A.; Boyko, M.; Shapira, Y.; Zlotnik, A. Blood glutamate scavenging: Insight into neuro protection. Int. J. Mol. Sci. 2012, 13, 10041–10066. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Maulucci-Gedde, M.; Kriegstein, A.R. Glutamate Neurotoxicity in Cortical Cell Culture. J. Neurosci. 1987, 7, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A.; Farrell, K.; Simon, R.P. Acidosis causes failure of astrocyte glutamate uptake during hypoxia. J. Cereb. Blood Flow Metab. 1995, 15, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Schurr, A.; Payne, R.S.; Rigor, B.M. Synergism between diltiazem and MK-801 but not APV in protecting hippocampal slices against hypoxic damage. Brain Res. 1995, 684, 233–236. [Google Scholar] [CrossRef]
- Husi, H.; Ward, M.A.; Choudhary, J.S.; Blackstock, W.P.; Grant, S.G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 2000, 3, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Husi, H. NMDA Receptors, Neural Pathways, and Protein Interaction Databases. Int. Rev. Neurobiol. 2004, 61, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.; Desalle, R.; Lam, H.; Meisel, L.; Coruzzi, G. Molecular Evolution of Glutamate Receptors: A Primitive Signaling Mechanism that Existed Before Plants and Animals Diverged. Mol. Biol. Evol. 1998, 16, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.J.; Grant, S.G.N. The origin and evolution of synapses. Nat. Rev. Neurosci. 2009, 10, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Yuzaki, M.; Aricescu, A.R. Review A GluD Coming-Of-Age Story. Trends Neurosci. 2017, 40, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca-B, P. Ectdomain shedding and regulated intracellular proteolysis in the central nervous system. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 337–359. [Google Scholar] [CrossRef]
- Montes de Oca, B. Pavel Metaloproteases in Adaptative Cell responses. In Proteases in Physiology and Pathology; Dhalla, S.C., Naranjan, S., Eds.; Springer: Singapore, 2017. [Google Scholar]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montes de Oca Balderas, P. Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities. Int. J. Mol. Sci. 2018, 19, 3800. https://doi.org/10.3390/ijms19123800
Montes de Oca Balderas P. Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities. International Journal of Molecular Sciences. 2018; 19(12):3800. https://doi.org/10.3390/ijms19123800
Chicago/Turabian StyleMontes de Oca Balderas, Pavel. 2018. "Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities" International Journal of Molecular Sciences 19, no. 12: 3800. https://doi.org/10.3390/ijms19123800
APA StyleMontes de Oca Balderas, P. (2018). Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities. International Journal of Molecular Sciences, 19(12), 3800. https://doi.org/10.3390/ijms19123800