Mutations Utilize Dynamic Allostery to Confer Resistance in TEM-1 β-lactamase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
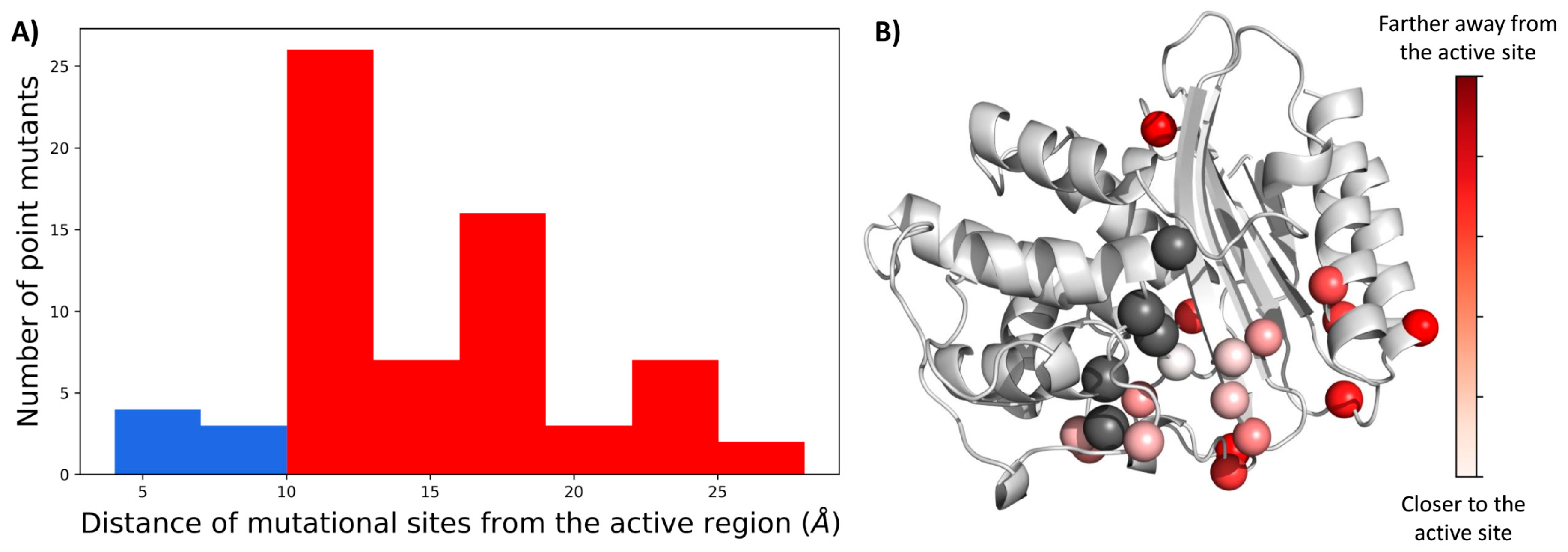
2.1. A Majority of the Resistance Driving Mutations Are Distal to the Active Site
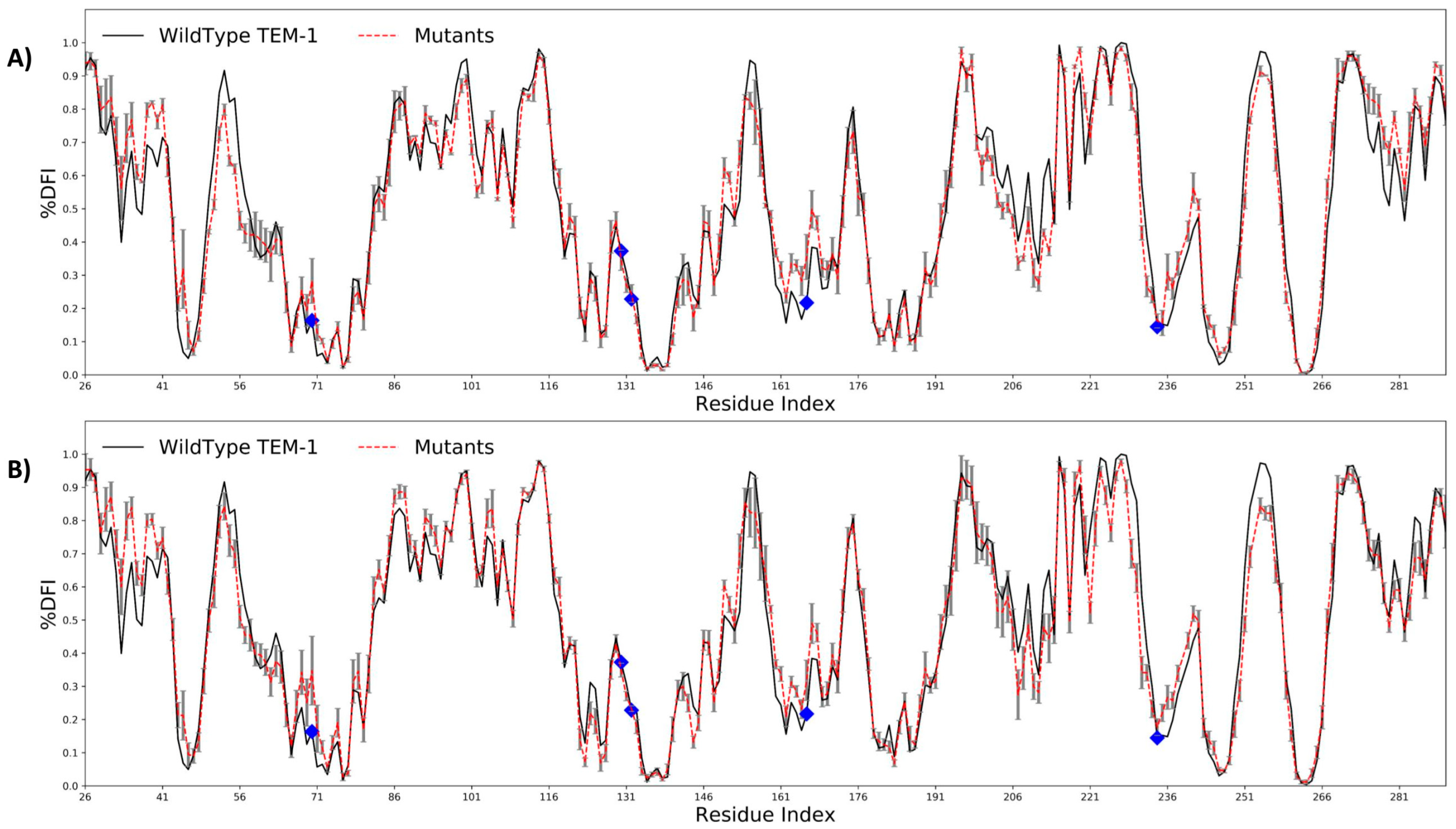
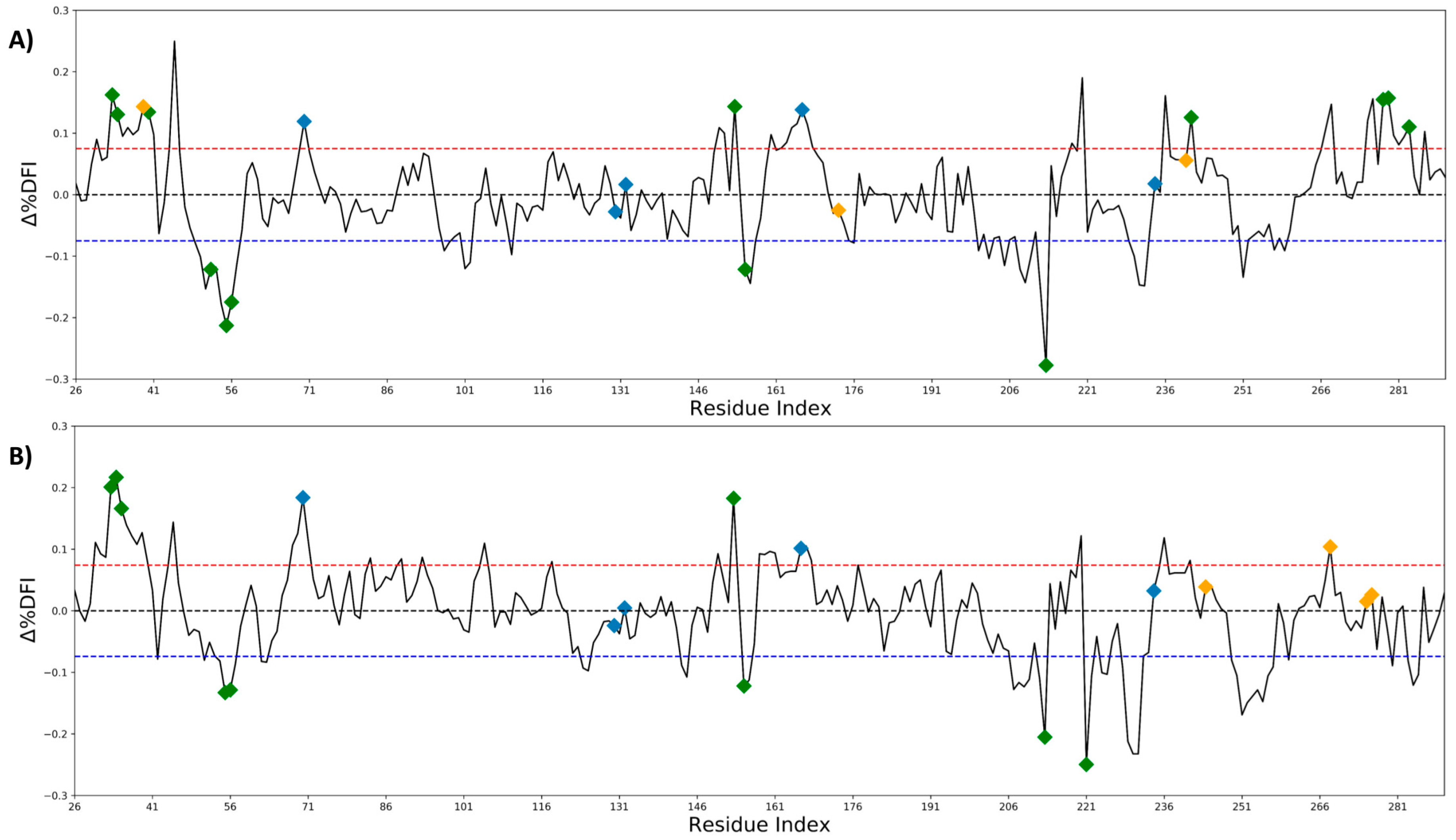
2.2. Antibiotic Resistance Driving Single Point Mutations Alter the Flexibility Profile of TEM-1 β-lactamase
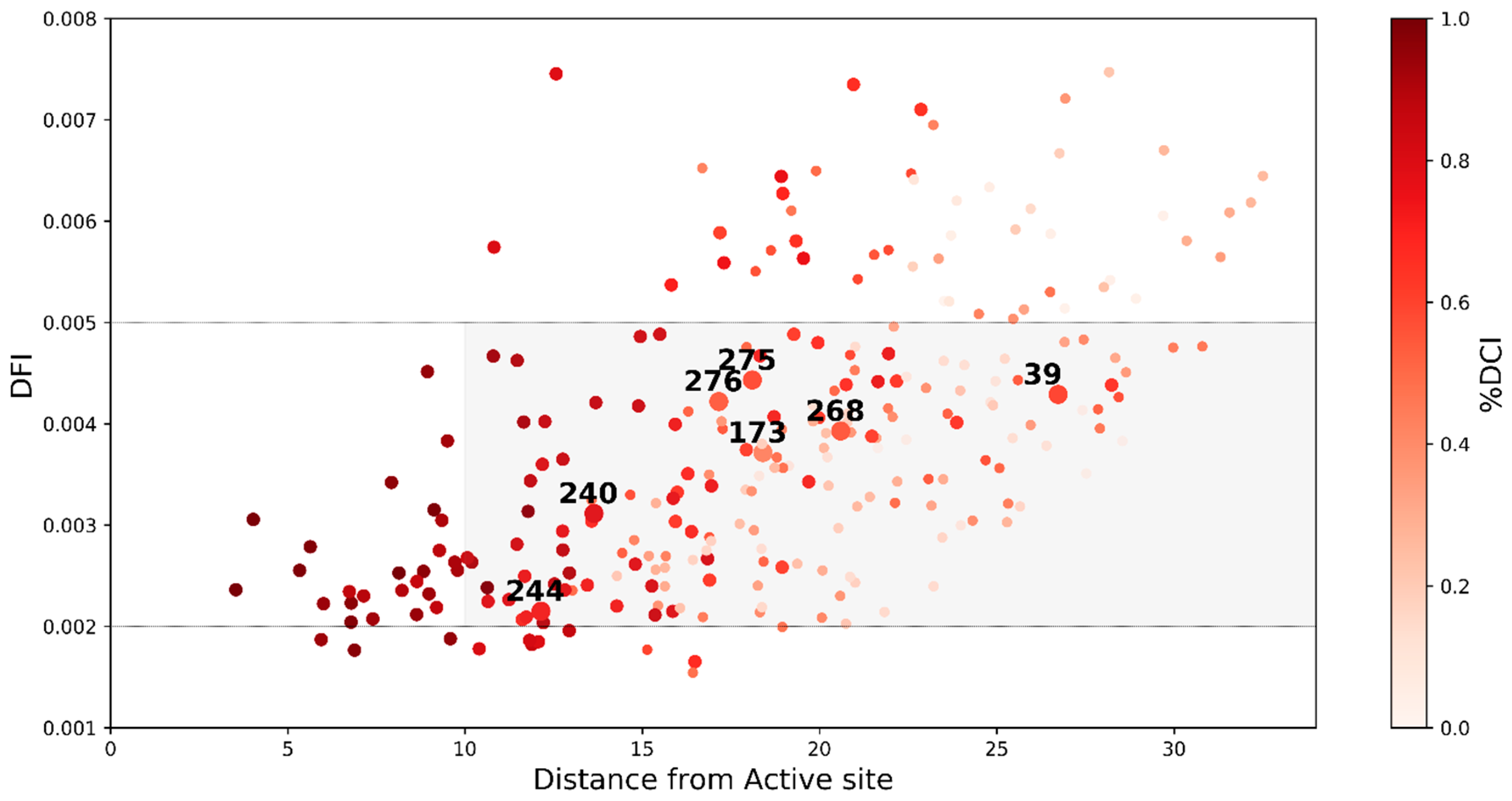
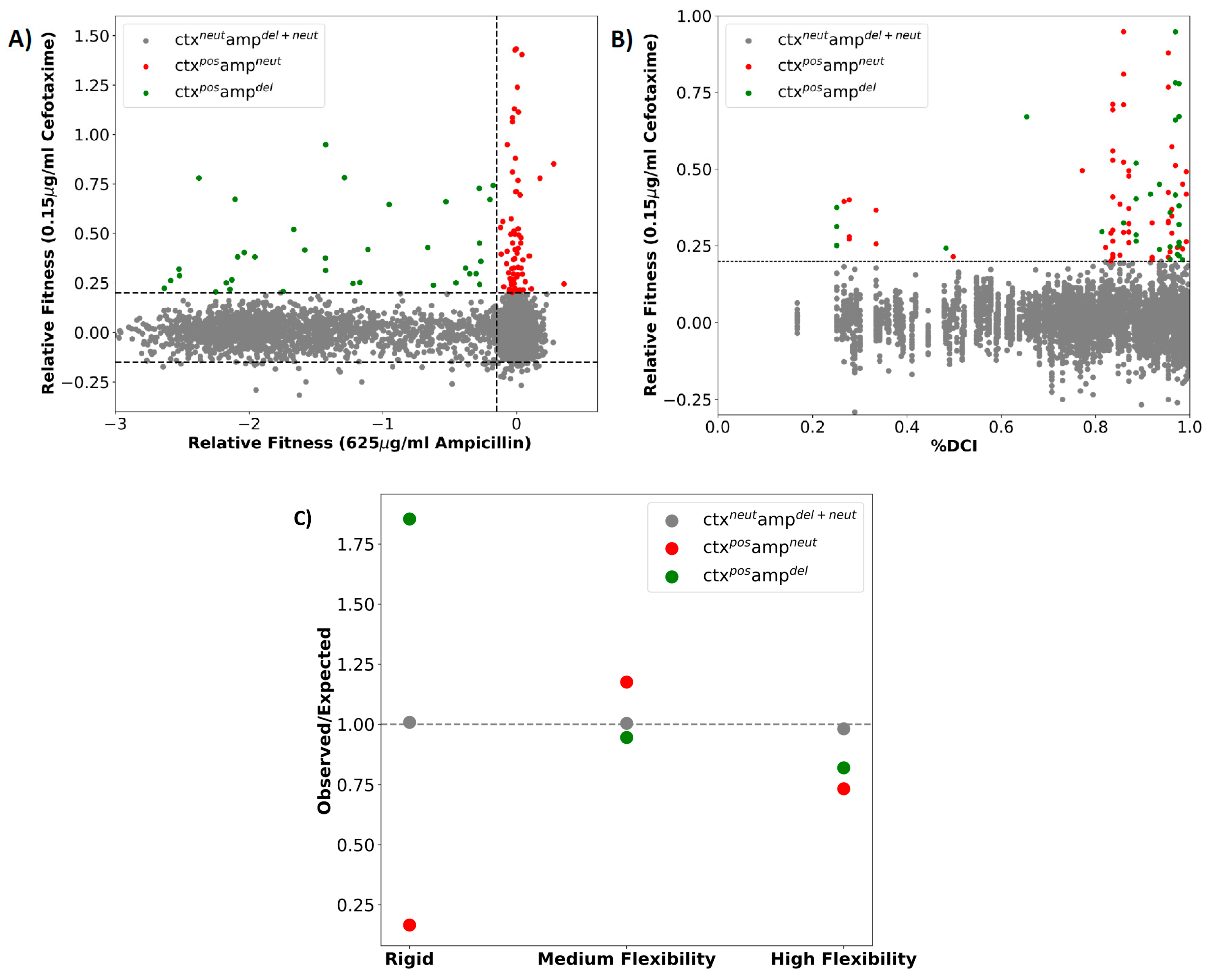
2.3. Dynamic Coupling Index (DCI) Gives an Insight into the Internal Network of Interactions in TEM-1 β-lactamase Protein
3. Methods
3.1. Dynamic Flexibility Index
3.2. Dynamic Coupling Index
3.3. Molecular Dynamic Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Appelbaum, P.C. 2012 and beyond: Potential for the start of a second pre-antibiotic era? J. Antimicrob. Chemother. 2012, 67, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Risso, V.A.; Gavira, J.A.; Sanchez-Ruiz, J.M.; Ozkan, S.B. Evolution of conformational dynamics determines the conversion of a promiscuous generalist into a specialist enzyme. Mol. Biol. Evol. 2015, 32, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Risso, V.A.; Gavira, J.A.; Mejia-Carmona, D.F.; Gaucher, E.A.; Sanchez-Ruiz, J.M. Hyperstability and substrate promiscuity in laboratory resurrections of Precambrian β-lactamases. J. Am. Chem. Soc. 2013, 135, 2899–2902. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.J.; Thornton, J.W. Analyzing protein structure and function using ancestral gene reconstruction. Curr. Opin. Struct. Biol. 2010, 20, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salverda, M.L.M.; De Visser, J.A.G.M.; Barlow, M. Natural evolution of TEM-1 β-lactamase: Experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 2010, 34, 1015–1036. [Google Scholar] [CrossRef] [PubMed]
- Ingles-Prieto, A.; Ibarra-Molero, B.; Delgado-Delgado, A.; Perez-Jimenez, R.; Fernandez, J.M.; Gaucher, E.A.; Sanchez-Ruiz, J.M.; Gavira, J.A. Conservation of protein structure over four billion years. Structure 2013, 21, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Darwinian Evolution Can Follow Only Very Few Mutational Paths to Fitter Proteins. Science. Available online: http://science.sciencemag.org/content/312/5770/111 (accessed on 1 October 2018).
- Raynes, Y.; Wylie, C.S.; Sniegowski, P.D.; Weinreich, D.M. Sign of selection on mutation rate modifiers depends on population size. Proc. Natl. Acad. Sci. USA 2018, 115, 3422–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreich, D.M.; Lan, Y.; Wylie, C.S.; Heckendorn, R.B. Should evolutionary geneticists worry about higher-order epistasis? Curr. Opin. Genet. Dev. 2013, 23, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knies, J.L.; Cai, F.; Weinreich, D.M. Enzyme Efficiency but Not Thermostability Drives Cefotaxime Resistance Evolution in TEM-1 β-Lactamase. Mol. Biol. Evol. 2017, 34, 1040–1054. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Serene, S.; Chao, H.X.; Gore, J. Hidden Randomness between Fitness Landscapes Limits Reverse Evolution. Phys. Rev. Lett. 2011, 106, 198102. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.R.; Geissler, P.L. Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. USA 2012, 109, 11681–11686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, M.I.; Hart, K.M.; Sibbald, C.A.; Frederick, T.E.; Jimah, J.R.; Knoverek, C.R.; Tolia, N.H.; Bowman, G.R. Prediction of New Stabilizing Mutations Based on Mechanistic Insights from Markov State Models. ACS Cent. Sci. 2017, 3, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Cortina, G.A.; Kasson, P.M. Predicting allostery and microbial drug resistance with molecular simulations. Curr. Opin. Struct. Biol. 2018, 52, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Cortina, G.A.; Kasson, P.M. Excess Positional Mutual Information Predicts Both Local and Allosteric Mutations Affecting Beta Lactamase Drug Resistance. Bioinformatics 2016, 32, 3420–3427. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.R.; Bolin, E.R.; Hart, K.M.; Maguire, B.C.; Marqusee, S. Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. USA 2015, 112, 2734–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, J.R.; Shoichet, B.K. Allosteric Inhibition Through Core Disruption. J. Mol. Biol. 2004, 336, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Modi, T.; Huihui, J.; Ghosh, K.; Ozkan, S.B. Ancient thioredoxins evolved to modern-day stability–function requirement by altering native state ensemble. Philos. Trans. R Soc. B 2018, 373, 20170184. [Google Scholar] [CrossRef] [PubMed]
- Stiffler, M.A.; Hekstra, D.R.; Ranganathan, R. Evolvability as a function of purifying selection in TEM-1 β-lactamase. Cell 2015, 160, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zou, T.; Modi, C.; Dörner, K.; Grunkemeyer, T.J.; Chen, L.; Fromme, R.; Matz, M.V.; Ozkan, S.B.; Wachter, R.M. A hinge migration mechanism unlocks the evolution of green-to-red photoconversion in GFP-like proteins. Structure 2015, 23, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Glembo, T.J.; Farrell, D.W.; Gerek, Z.N.; Thorpe, M.F.; Ozkan, S.B. Collective Dynamics Differentiates Functional Divergence in Protein Evolution. PLoS Comput. Biol. 2012, 8, e1002428. [Google Scholar] [CrossRef] [PubMed]
- Nevin Gerek, Z.; Kumar, S.; Banu Ozkan, S. Structural dynamics flexibility informs function and evolution at a proteome scale. Evol. Appl. 2013, 6, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, B.M.; Gerek, Z.N.; Kumar, S.; Ozkan, S.B. Conformational dynamics of nonsynonymous variants at protein interfaces reveals disease association. Proteins Struct. Funct. Bioinform. 2015, 83, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, J.L.; Baquero, F.; Andersson, D.I. Predicting antibiotic resistance. Nat. Rev. Microbiol. 2007, 5, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerek, Z.N.; Ozkan, S.B. Change in Allosteric Network Affects Binding Affinities of PDZ Domains: Analysis through Perturbation Response Scanning. PLoS Comput. Biol. 2011, 7, e1002154. [Google Scholar] [CrossRef] [PubMed]
- Gerek, Z.N.; Keskin, O.; Ozkan, S.B. Identification of specificity and promiscuity of PDZ domain interactions through their dynamic behavior. Proteins 2009, 77, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Stricher, F.; Serrano, L.; Tawfik, D.S. How Protein Stability and New Functions Trade Off. PLoS Comput. Biol. 2008, 4, e1000002. [Google Scholar] [CrossRef] [PubMed]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. Antibiotic tolerance facilitates the evolution of resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Trebosc, V.; Kemmer, C.; Rosenstiel, P.; Beardmore, R.; Schulenburg, H.; Jansen, G. Alternative Evolutionary Paths to Bacterial Antibiotic Resistance Cause Distinct Collateral Effects. Mol. Biol. Evol. 2017, 34, 2229–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figliuzzi, M.; Jacquier, H.; Schug, A.; Tenaillon, O.; Weigt, M. Coevolutionary Landscape Inference and the Context-Dependence of Mutations in Beta-Lactamase TEM-1. Mol. Biol. Evol. 2016, 33, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Cusack, M.P.; Thibert, B.; Bredesen, D.E.; Rio, G. del Efficient Identification of Critical Residues Based Only on Protein Structure by Network Analysis. PLoS ONE 2007, 2, e421. [Google Scholar] [CrossRef] [PubMed]
- Otten, R.; Liu, L.; Kenner, L.R.; Clarkson, M.W.; Mavor, D.; Tawfik, D.S.; Kern, D.; Fraser, J.S. Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat. Commun. 2018, 9, 1314. [Google Scholar] [CrossRef] [PubMed]
- Bershtein, S.; Segal, M.; Bekerman, R.; Tokuriki, N.; Tawfik, D.S. Robustness–epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 2006, 444, 929. [Google Scholar] [CrossRef] [PubMed]
- Miton, C.M.; Jonas, S.; Fischer, G.; Duarte, F.; Mohamed, M.F.; van Loo, B.; Kintses, B.; Kamerlin, S.C.L.; Tokuriki, N.; Hyvönen, M.; et al. Evolutionary repurposing of a sulfatase: A new Michaelis complex leads to efficient transition state charge offset. Proc. Natl. Acad. Sci. USA 2018, 115, E7293–E7302. [Google Scholar] [CrossRef] [PubMed]
- Keedy, D.A.; Hill, Z.B.; Biel, J.T.; Kang, E.; Rettenmaier, T.J.; Brandão-Neto, J.; Pearce, N.M.; von Delft, F.; Wells, J.A.; Fraser, J.S. An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.C.; Correy, G.J.; Mabbitt, P.D.; Buckle, A.M.; Tokuriki, N.; Jackson, C.J. Laboratory evolution of protein conformational dynamics. Curr. Opin. Struct. Biol. 2018, 50, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Butler, B.M.; Kumar, S.; Ozkan, S.B. Integration of structural dynamics and molecular evolution via protein interaction networks: A new era in genomic medicine. Curr. Opin. Struct. Biol. 2015, 35, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Larrimore, K.E.; Kazan, I.C.; Kannan, L.; Kendle, R.P.; Jamal, T.; Barcus, M.; Bolia, A.; Brimijoin, S.; Zhan, C.-G.; Ozkan, S.B.; et al. Plant-expressed cocaine hydrolase variants of butyrylcholinesterase exhibit altered allosteric effects of cholinesterase activity and increased inhibitor sensitivity. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Glembo, T.J.; Ozkan, S.B. The Role of Conformational Dynamics and Allostery in the Disease Development of Human Ferritin. Biophys. J. 2015, 109, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Ikeguchi, M.; Ueno, J.; Sato, M.; Kidera, A. Protein structural change upon ligand binding: Linear response theory. Phys. Rev. Lett. 2005, 94, 078102. [Google Scholar] [CrossRef] [PubMed]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Bastolla, U. Computing protein dynamics from protein structure with elastic network models. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 488–503. [Google Scholar] [CrossRef]
- Zheng, W.; Brooks, B.R.; Thirumalai, D. Allosteric transitions in biological nanomachines are described by robust normal modes of elastic networks. Curr. Protein Pept. Sci. 2009, 10, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Tirion, M.M. Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Yang, L.-W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Jelsch, C.; Mourey, L.; Masson, J.M.; Samama, J.P. Crystal structure of Escherichia coli TEM1 beta-lactamase at 1.8 A resolution. Proteins 1993, 16, 364–383. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1. (2010); Schrödinger: Portland, OR, USA, 2010. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Neria, E.; Fischer, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles; Taylor & Francis, Inc.: Bristol, PA, USA, 1988; ISBN 978-0-85274-392-8. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Vakulenko, S.B.; Geryk, B.; Kotra, L.P.; Mobashery, S.; Lerner, S.A. Selection and characterization of beta-lactam-beta-lactamase inactivator-resistant mutants following PCR mutagenesis of the TEM-1 beta-lactamase gene. Antimicrob. Agents Chemother. 1998, 42, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- McLeish, T.C.B.; Cann, M.J.; Rodgers, T.L. Dynamic Transmission of Protein Allostery without Structural Change: Spatial Pathways or Global Modes? Biophys. J. 2015, 109, 1240–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Modi, T.; Ozkan, S.B. Mutations Utilize Dynamic Allostery to Confer Resistance in TEM-1 β-lactamase. Int. J. Mol. Sci. 2018, 19, 3808. https://doi.org/10.3390/ijms19123808
Modi T, Ozkan SB. Mutations Utilize Dynamic Allostery to Confer Resistance in TEM-1 β-lactamase. International Journal of Molecular Sciences. 2018; 19(12):3808. https://doi.org/10.3390/ijms19123808
Chicago/Turabian StyleModi, Tushar, and S. Banu Ozkan. 2018. "Mutations Utilize Dynamic Allostery to Confer Resistance in TEM-1 β-lactamase" International Journal of Molecular Sciences 19, no. 12: 3808. https://doi.org/10.3390/ijms19123808
APA StyleModi, T., & Ozkan, S. B. (2018). Mutations Utilize Dynamic Allostery to Confer Resistance in TEM-1 β-lactamase. International Journal of Molecular Sciences, 19(12), 3808. https://doi.org/10.3390/ijms19123808