An Animal Model for Assessing the Effects of Hydroxyurea Exposure Suggests That the Administration of This Agent to Pregnant Women and Young Infants May Not Be as Safe as We Thought

Abstract
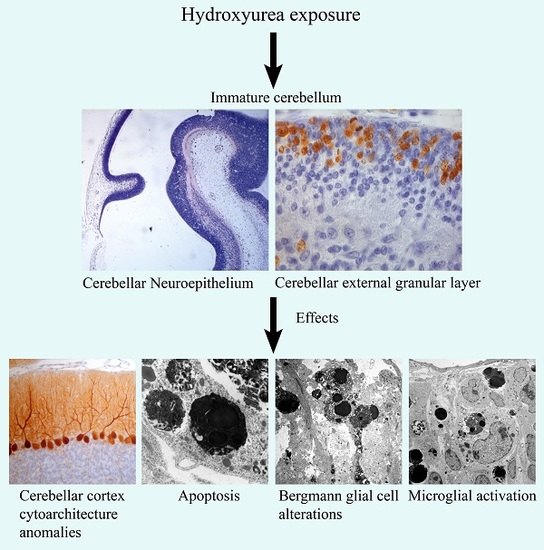
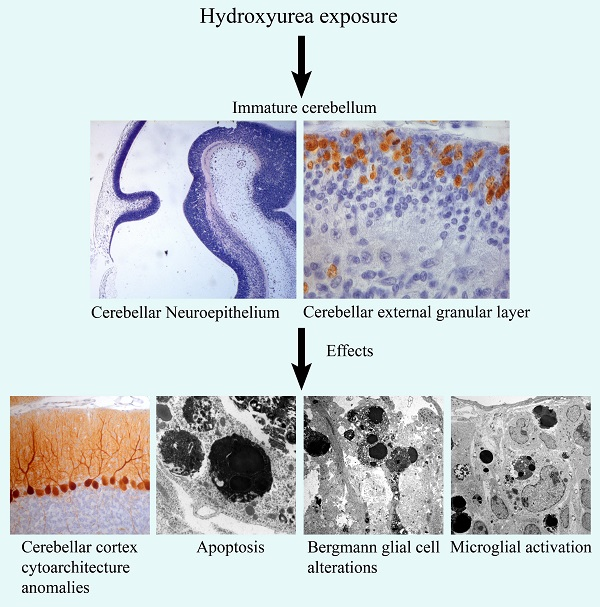
:
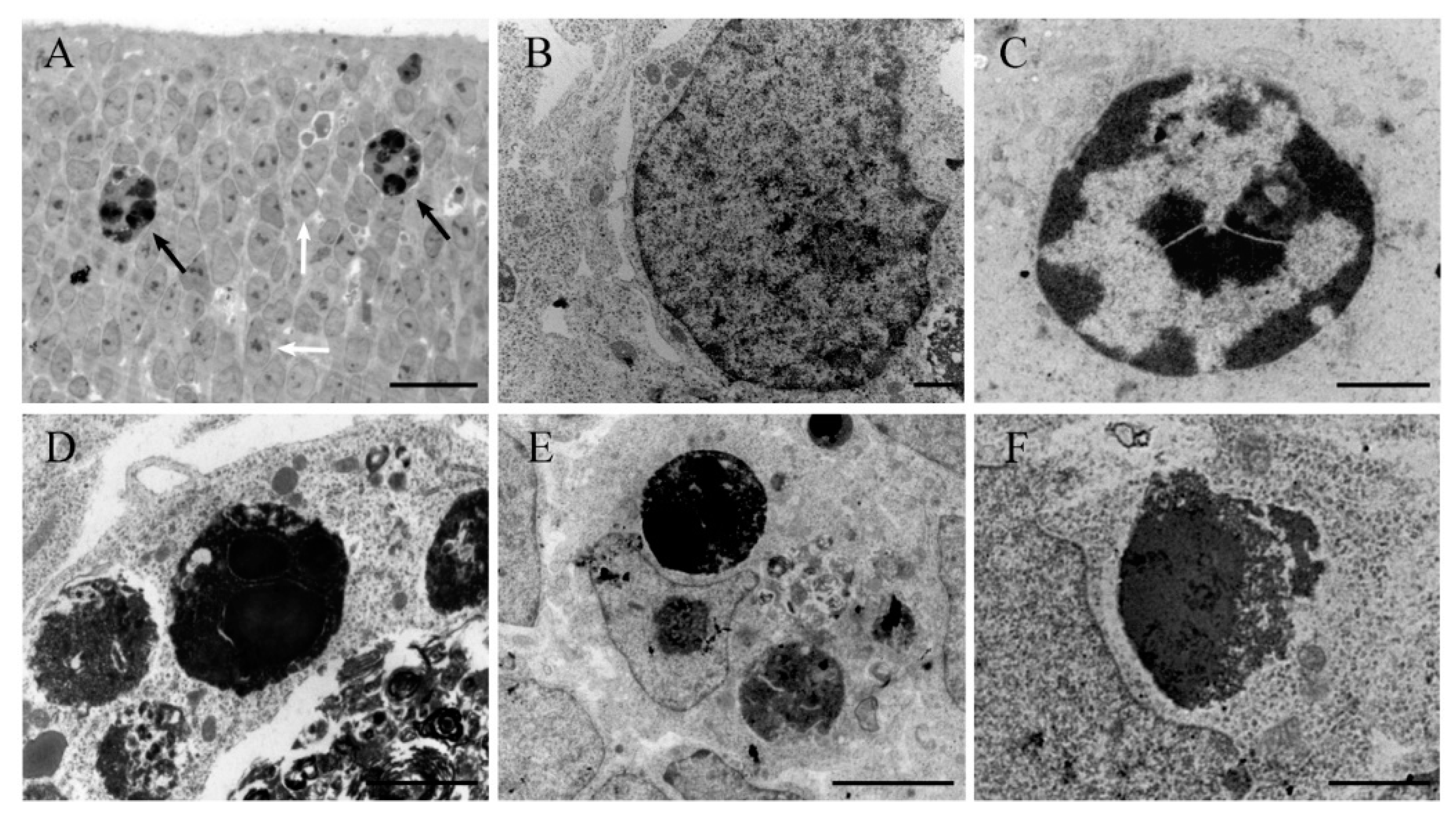
{kind=link}
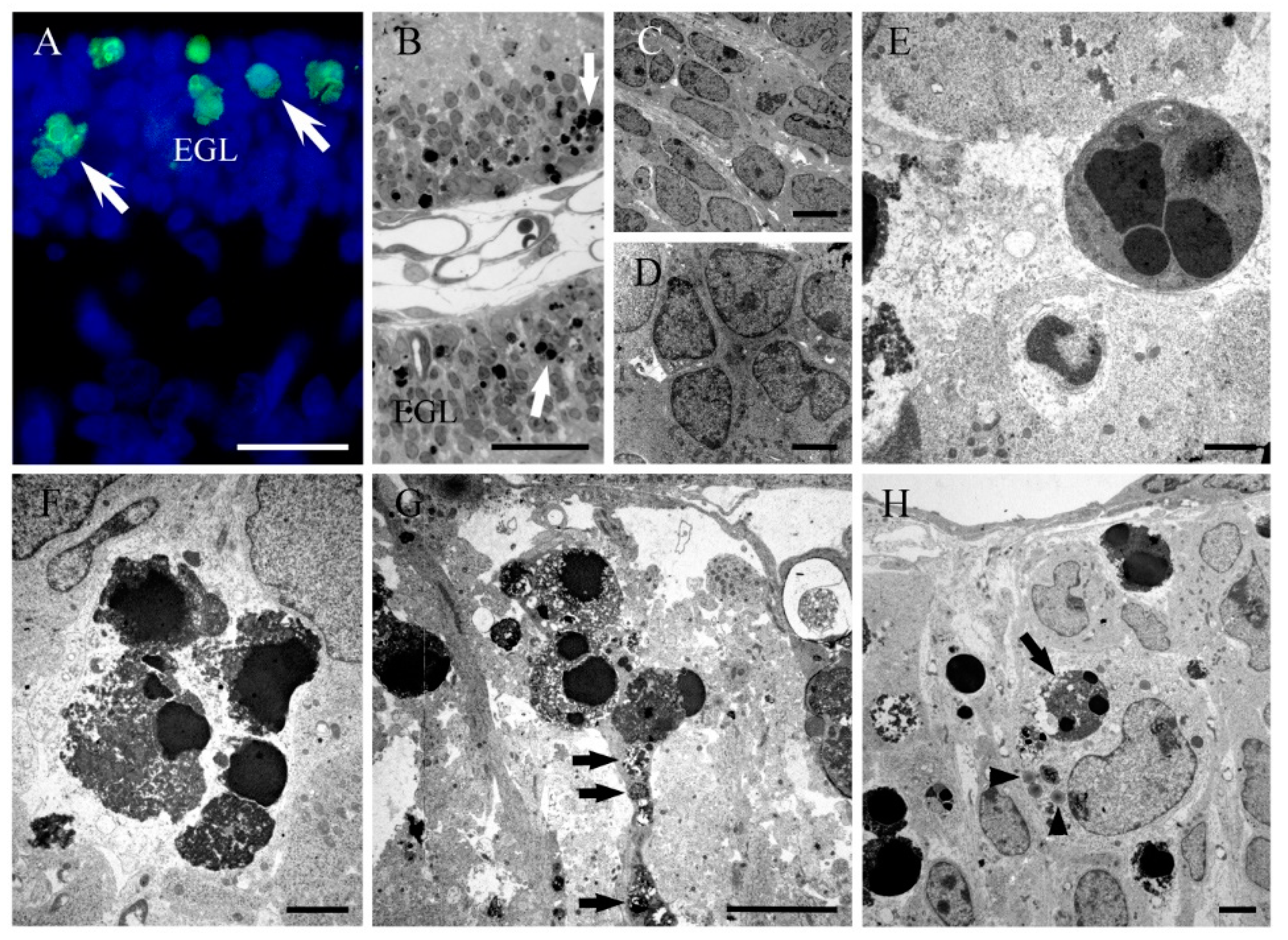
{kind=link}
{kind=link}
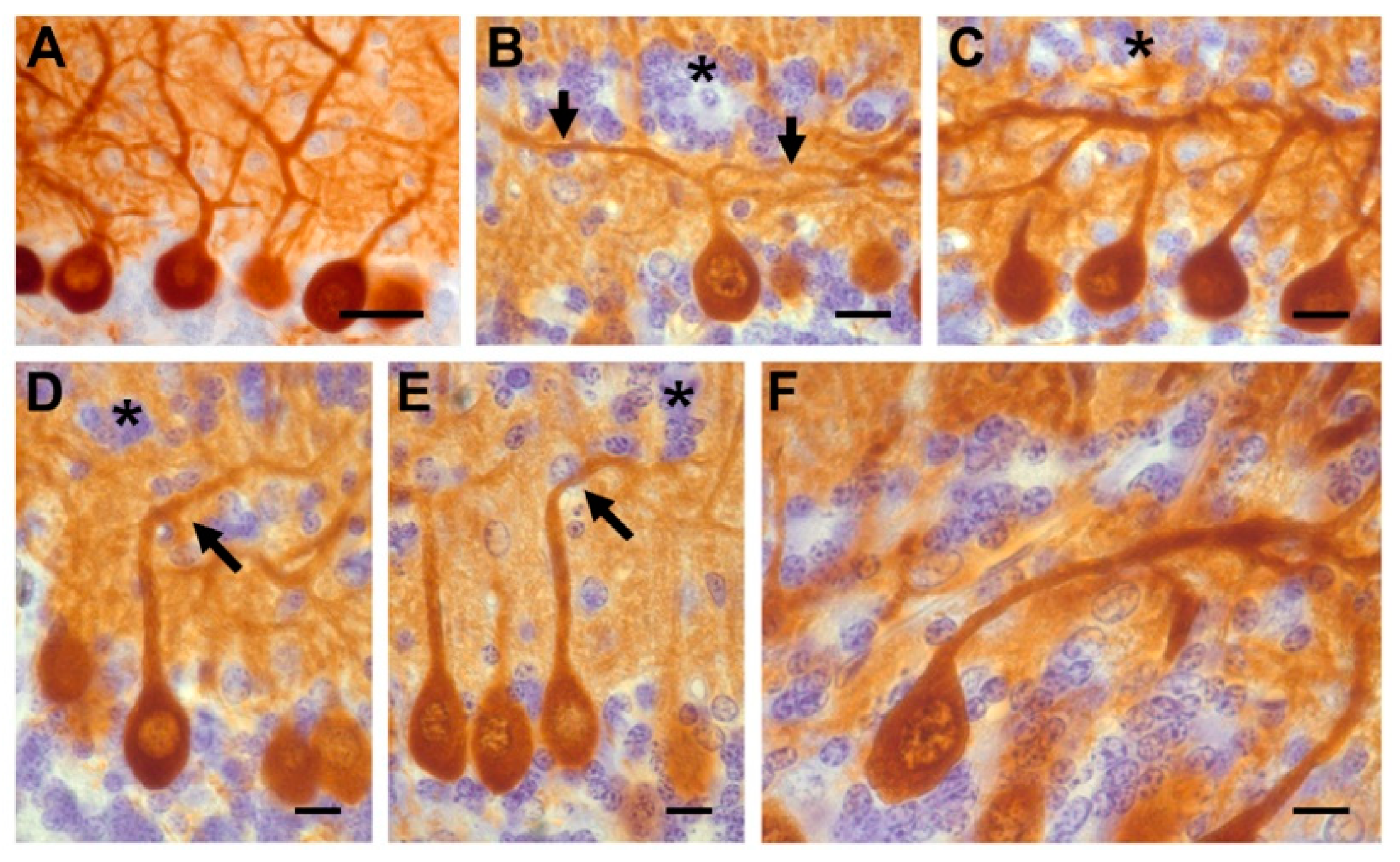{kind=link}
1. Introduction
2. Hydroxyurea: An Overview
3. Mechanisms of Action of the Hydroxyurea
4. Teratogenic Effects of Hydroxyurea
5. Justifying the Choice of the Cerebellum as a Model to Assess the Effects of Hydroxyurea Exposure
6. Embryonic Effects of HU Exposure: Short-Survival Experiments
7. Embryonic Effects of HU Exposure: Long-Survival Experiments
8. Perinatal Effects of HU Exposure: Short-Survival Experiments
9. Perinatal Effects of HU Exposure: Long-Survival Experiments
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BrdU | 5-bromo-2′-deoxyuridine |
| DCN | Deep cerebellar nuclei |
| E | Embryonic day |
| EGL | External granular layer |
| GCs | Granule cells |
| GL | Granular layer |
| HU | Hydroxyurea |
| ML | Molecular layer |
| P | Postnatal day |
| PCs | Postnatal day |
References
- Koç, A.; Wheeler, L.J.; Mathews, C.K.; Merrill, G.F. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J. Biol. Chem. 2004, 279, 223–230. [Google Scholar] [CrossRef]
- McGann, P.T.; Ware, R.E. Hydroxyurea therapy for sickle cell anemia. Expert Opin. Drug Saf. 2015, 14, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levorino, L.G.; Baldin, P.E.; Picado, S.M.; Calil, K.B.; Viel, A.A.; Campos, L.A. Prevalence of sickle cell disease and sickle cell trait in national neonatal screening studies. Rev. Bras. Hematol. Hemoter. 2011, 33, 49–54. [Google Scholar] [CrossRef]
- De Montalembert, M.; Belloy, M.; Bernaudin, F.; Gouraud, F.; Capdeville, R.; Mardini, R.; Philippe, N.; Jais, J.P.; Bardakdjian, J.; Ducrocq, R.; et al. Three-year follow-up of hydroxyurea treatment in severely ill children with sickle cell disease. The French Study Group on Sickle Cell Disease. J. Pediatr. Hematol. Oncol. 1997, 19, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.; Archibong, A.E.; Powell, A.; Strange, B.; Roberson, S.; Hills, E.R.; Bourne, P. Perturbation of the developmental potential of preimplantation mouse embryos by hydroxyurea. Int. J. Environ. Res. Public Health 2010, 7, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Strouse, J.J.; Lanzkron, S.; Beach, M.C.; Haywood, C.; Park, H.; Witkop, C.; Wilson, R.F.; Bass, E.B.; Segal, J.B. Hydroxyurea for sickle cell disease: A systematic review for efficacy and toxicity in children. Pediatrics 2008, 122, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Hankins, J.S.; Ware, R.E.; Rogers, Z.R.; Wynn, L.W.; Lane, P.A.; Scott, J.P.; Wang, W.C. Long-term hydroxyurea therapy for infants with sickle cell anemia: The HUSOFT extension study. Blood 2005, 106, 2269–2275. [Google Scholar] [CrossRef] [PubMed]
- Woo, G.H.; Katayama, K.; Jung, J.Y.; Uetsuka, K.; Bak, E.J.; Nakayama, H.; Doi, K. Hydroxyurea (HU)-induced apoptosis in the mouse fetal tissues. Histol. Histopathol. 2003, 18, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Woo, G.H.; Bak, E.J.; Katayama, K.; Doi, K. Molecular mechanisms of hydroxyurea (HU)-induced apoptosis in the mouse fetal brain. Neurotoxicol. Teratol. 2006, 28, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Ebels, E.J.; Peters, I.; Thijs, A. Studies on ectopic granule cells in the cerebellar cortex. III. An investigation into the restoration of the external granular layer after partial destruction. Acta Neuropathol. 1975, 31, 103–107. [Google Scholar] [CrossRef]
- Koppel, H.; Lewis, P.D.; Padel, A.J. Cell death in the external granular layer of normal and undernourished rats: Futher observations, including estimates of rate of cell loss. Cell Tissue Kinet. 1983, 16, 99–106. [Google Scholar]
- Navarra, P.; Preziosi, P. Hydroxyurea: New insights on an old drug. Crit. Rev. Oncol. Hematol. 1999, 29, 249–255. [Google Scholar] [CrossRef]
- Dresler, W.F.C.; Stein, R. Ueber den Hydroxylharnstoff. Ann. Chem. Pharmacol. 1869, 150, 242–252. [Google Scholar] [CrossRef]
- Newton, H.B. Hydroxyurea chemotherapy in the treatment of meningiomas. Neurosurg. Focus 2007, 23, E11. [Google Scholar] [CrossRef] [Green Version]
- Saban, N.; Bujak, M. Hydroxyurea and hydroxamic acid derivatives as antitumor drugs. Cancer Chemother. Pharmacol. 2009, 64, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; Depotovic, J.M.; Mortier, N.A.; Flanagan, J.M.; He, J.; Smeltzer, M.P.; Kimble, A.C.; Aygun, B.; Wu, S.; Howard, T.; et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood 2011, 118, 4985–4991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebwohl, M.; Menter, A.; Koo, J.; Feldman, S.R. Combination therapy to treat moderate to severe psoriasis. J. Am. Acad. Dermatol. 2004, 50, 416–430. [Google Scholar] [CrossRef]
- Lori, F.; Kelly, L.M.; Foli, A.; Lisziewicz, J. Safety of hydroxyurea in the treatment of HIV infection. Expert Opin. Drug Saf. 2004, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Model List of Essential Medicines; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Madaan, K.; Kaushik, D.; Verma, T. Hydroxyurea: A key player in cancer chemotherapy. Expert Rev. Anticancer Ther. 2012, 12, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Borenfreund, E.; Krimm, M.; Bendich, A. Chromosomal aberrations induced by hyponitrite and hydroxylamine derivates. J. Natl. Cancer Inst. 1964, 32, 667–679. [Google Scholar] [PubMed]
- Weinlich, G.; Fritsch, P. Leg ulcers in patients treated with hydroxyurea for myeloproliferative disorders: What is the trigger? Br. J. Dermatol. 1999, 141, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.; Cantwell, B.M.; Kerr, N.; Upfold, A.; Harris, A.L. Phase 1 study of high-dose hydroxyurea in lung cancer. Cancer Chemother. Pharmacol. 1988, 21, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.W.; Kohanski, M.A.; Simmons, L.A.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol. Cell 2009, 36, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Zhou, B.; Chu, B.; Yen, Y. Ribonucleotide reductase inhibitors and future drug desing. Curr. Cancer Drug Targets 2006, 6, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Mannargudi, M.B.; Deb, S. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: Is it a viable cancer theraphy? J. Cancer Res. Oncol. 2017, 143, 1499–1529. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, N.; Schlisser, A.E.; Hales, B.F. Editor’s highlight: Hydroxyurea exposure activates the P53 signaling pathway in murine organogenesis-stage embryos. Toxicol. Sci. 2016, 152, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.E.; Shook, L.M.; Ware, R.E.; Brinkman, W.B. Shared decision making for hydroxyurea treatment initiation in children with sickle cell anemia. Pedriatr. Blood Cancer 2015, 62, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Thauvin-Robinet, C.; Maingueneau, C.; Robert, E.; Elefant, E.; Guy, H.; Caillot, D.; Casasnovas, R.O.; Douvier, S.; Nivelon-Chevallier, A. Exposure to hydroxyurea during pregnancy: A case series. Leukemia 2001, 15, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K.; McCarthy, W.F.; Guo, N.; DeCastro, L.; Bellevue, R.; Barton, B.A.; Waclawiw, M.A. Multicenter study of hydroxyurea in sickle cell. J. Natl. Med. Assoc. 2009, 101, 1046–1051. [Google Scholar] [CrossRef]
- Lanzkron, S.; Strouse, J.J.; Wilson, R.; Beach, M.C.; Haywood, C.; Park, H.; Witkop, C.; Bass, E.B.; Segal, J.B. Systematic review: Hydroxyurea for the treatment of adults with sickle cell disease. Ann. Intern. Med. 2008, 148, 939–955. [Google Scholar] [CrossRef] [PubMed]
- Woo, G.H.; Bak, E.J.; Katayama, K.; Doi, K. Hydroxyurea (HU)-induced apoptosis in the mouse fetal lung. Exp. Mol. Pathol. 2005, 79, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Schlisser, A.E.; Hales, B.F. Deprenyl enhances the teratogenicity of hydroxyurea in organogenesis stage mouse embryos. Toxicol. Sci. 2013, 134, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Banh, S.; Hales, B.F. Hydroxyurea exposure triggers tissue-specific activation of p38 mitogen-activated protein kinase signaling and the DNA damage response inorganogenesis-stage mouse embryos. Toxicol. Sci. 2013, 133, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Woo, G.H.; Katayama, K.; Bak, E.J.; Ueno, H.; Tamauchi, H.; Uetsuka, K.; Nakayama, H.; Doi, K. Effects of prenatal hydroxyurea-treatment on mouse offspring. Exp. Toxicol. Pathol. 2004, 56, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.; Bayer, S. Development of the Cerebellar System. In Relation to its Evolution, Structure and Functions; CRC Press, Inc.: Boca Raton, FL, USA, 1997. [Google Scholar]
- Goldowitz, D.; Hamre, K. The cells and molecules that make a cerebellum. Trends Neurosci. 1988, 21, 375–382. [Google Scholar] [CrossRef]
- Schilling, K.; Oberdick, J.; Rossi, F.; Baader, S.L. Besides Purkinje cells and granule neurons: An appraisal of the cell biology of the interneurons of the cerebellar cortex. Histochem. Cell. Biol. 2008, 130, 601–615. [Google Scholar] [CrossRef]
- Chetodal, A. Should I stay or should I go? Becoming a granule cell. Trends Neurosci. 2010, 33, 163–172. [Google Scholar] [CrossRef]
- Sillitoe, R.V.; Joyner, A.L. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu. Rev. Cell Dev. Biol. 2007, 23, 549–577. [Google Scholar] [CrossRef]
- Wullimann, M.F.; Mueller, T.; Distel, M.; Babaryka, A.; Grothe, B.; Köster, R.W. The long adventurous journey of rhombic lip in jawed vertebrates: A comparative developmental analysis. Front. Neuroanat. 2011, 5, 27. [Google Scholar] [CrossRef]
- Leto, K.; Rolando, C.; Rossi, F. The genesis of cerebellar GABAergic neurons: Fate potential and specification mechanisms. Front. Neuroanat. 2012, 6, 6. [Google Scholar] [CrossRef]
- Manto, M.; Marmolino, M. Cerebellar disorders—At the crossroad of molecular pathways and diagnosis. Cerebellum 2009, 8, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Manto, M. Toxic agents causing cerebellar ataxias. Handb. Clin. Neurol. 2012, 103, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Koning, I.V.; Tielemans, M.J.; Hoebeek, F.E.; Ecury-Goossen, G.M.; Reiss, I.K.M.; Steegers-Theunissen, R.P.M.; Dudink, J. Impacts on prenatal development of the human cerebellum: A systematic review. J. Matern. Fetal Neonatal Med. 2017, 30, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Ladonde, R.; Strazielle, C. Spontaneous and induced mouse mutations with cerebellar dysfunctions: Behavior and neurochemistry. Brain Res. 2007, 1140, 51–74. [Google Scholar] [CrossRef] [PubMed]
- Martí, J.; Santa-Cruz, M.C.; Bayer, S.A.; Ghetti, B.; Hervás, J.P. Purkinje cell age-distribution in fissures and in foliar crowns: A comparative study in the weaver cerebellum. Brain Struct. Funct. 2007, 212, 347–357. [Google Scholar] [CrossRef]
- Martí, J.; Santa-Cruz, M.C.; Serra, R.; Hervás, J.P. Hydroxyurea treatment and development of the rat cerebellum: Effects on the neurogenetic profiles and settled patterns of Purkinje cells and deep cerebellar nuclei neurons. Neurotox. Res. 2016, 30, 563–580. [Google Scholar] [CrossRef]
- Bernocchi, G.; Bottone, M.G.; Piccolini, V.M.; Dal Bo, V.; Santin, G.; De Pascali, S.A.; Migoni, D.; Fanizzi, F.P. Developing central nervous system and vulnerability to platinum compounds. Chemther. Res. Pract. 2011, 315418. [Google Scholar] [CrossRef] [PubMed]
- Bottone, M.G.; Veronica, D.B.; Piccolini, V.M.; Bottiroli, G.; De Pascali, S.A.; Fanizzi, F.P.; Bernocchi, G. Developmental expression of cellular prion protein and apoptotic molecules in the rat cerebellum: Effects of platinum compounds. J. Chem. Neuroanat. 2012, 46, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, O.M.; Abd El-Tawab, S.M.; Ahmed, R.G. Effects of experimentally induced maternal hypothyroidism and hyperthyroidism on the development of rat offspring: I. The development of the thyroid hormones-neurotransmitters and adenosinergic system interactions. Int. J. Dev. Neurosci. 2010, 28, 437–454. [Google Scholar] [CrossRef]
- Luo, J. Mechanisms of ethanol-induced death of cerebellar granule cells. Cerebellum 2012, 11, 145–154. [Google Scholar] [CrossRef]
- Oster-Granite, M.L.; Herndon, R.M. The pathogenesis of parvovirus-induced cerebellar hypoplasia in the Syrian hamster, Mesocricetus auratus. Fluorescent antibody, foliation, cytoarchitecture, Golgi and electron microscopic studies. J. Comp. Neurol. 1976, 169, 481–521. [Google Scholar] [CrossRef]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluropotent stem cells. Cell Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vázquez, L.; Martí, J. Effects of hydroxyurea exposure on the rat cerebellar neuroepithelium: An immunohistochemical and electron microscopic study along the anteroposterior and mediolateral axes. Neurotox. Res. 2017, 32, 671–682. [Google Scholar] [CrossRef]
- Martí, J.; Santa-Cruz, M.C.; Serra, R.; Hervás, J.P. Systematic differences in timeof cerebellar-neuron origin derived from bromodeoxyuridine immunoperoxidase staining protocols and tritiated thymidine autoradiography: A comparative study. Int. J. Dev. Neurosci. 2015, 47, 216–228. [Google Scholar] [CrossRef]
- Kerr, J.F.R. Neglected opportunities in apoptosis research. Trends Cell Biol. 1995, 5, 55–57. [Google Scholar] [CrossRef]
- Butts, T.; Green, M.J.; Wingate, R.J. Development of the cerebellum: Simple steps to make a “little brain”. Development 2014, 141, 4031–4041. [Google Scholar] [CrossRef] [PubMed]
- Marzban, H.; Del Bigio, M.R.; Alizadeh, J.; Ghavami, S.; Zachariah, R.M.; Rastegar, M. Cellular commitment in the developing cerebellum. Front. Cell Neurosci. 2015, 8, 450. [Google Scholar] [CrossRef]
- Sekerkova, G.; Ilijic, E.; Mugnaini, E. Time of origin of unipolar brush cells in the rat cerebellum as observed by prenatal bromodeoxyuridine labeling. Neuroscience 2004, 127, 845–858. [Google Scholar] [CrossRef]
- Martí, J.; Molina, V.; Santa-Cruz, M.C.; Hervás, J.P. Developmental injury to the cerebellar cortex following hydroxyurea treatment in early postnatal life: An immunohistochemical and electron microscopic study. Neurotox. Res. 2017, 31, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vázquez, L.; Vons, O.; Valero, O.; Martí, J. Hydroxyurea exposure and development of the cerebellar external granular layer: Effects on granule cell precursors, bergmann glial and microglial cells. Neurotox. Res. 2018. [Google Scholar] [CrossRef]
- Scheuer, T.; Sharkovska, Y.; Tarabykin, V.; Marggraf, K.; Brockmöller, V.; Bührer, C.; Endesfelder, S.; Schmitz, T. Neonatal Hyperoxia Perturbs Neuronal Development in the Cerebellum. Mol. Neurobiol. 2018, 55, 3901–3915. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Bostrom, M.; Ek, C.J.; Li, T.; Xie, C.; Xu, Y.; Sun, Y.; Blomgren, K.; Zhu, C. Radiation induces progenitor cell death, microglia activation, and blood-brain barrier damage in the juvenile rat cerebellum. Sci. Rep. 2017, 7, 46181. [Google Scholar] [CrossRef] [PubMed]
- Mousa, A.M.; Al-Fadhli, A.S.; Rao, M.S.; Kilarkaje, N. Gestational lead exposure induces developmental abnormalities and up-regulates apoptosis of fetal cerebellar cells in rats. Drug Chem. Toxicol. 2015, 38, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Taranukhin, A.G.; Taranukhina, E.Y.; Saransaari, P.; Pelto-Huikko, M.; Podkletnova, I.M.; Oja, S.S. Taurine protects cerebellar neurons of the external granular layer against ethanol-induced apoptosis in 7-day-old mice. Amino Acids 2012, 43, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, M.; Derry, W.B. Your neighbours matter–non-autonomous control of apoptosis in developmental and disease. Cell Death Differ. 2016, 23, 1110–1118. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease-a double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Stoodley, C.J. The cerebellum and neurodevelopmental disorders. Cerebellum 2016, 15, 34–37. [Google Scholar] [CrossRef]
- Bruchhage, M.M.K.; Bucci, M.P.; Becker, E.B.E. Cerebellar involvement in autism and ADHD. In Handbook of Clinical Neurology, 3rd ed.; Manto, M., Huisman, T.A.G.M., Eds.; The Cerebellum: Disorders and Treatment; Elsevier: Amsterdam, The Netherlands, 2003; Volume 155. [Google Scholar]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Vázquez, L.; Martí, J. An Animal Model for Assessing the Effects of Hydroxyurea Exposure Suggests That the Administration of This Agent to Pregnant Women and Young Infants May Not Be as Safe as We Thought. Int. J. Mol. Sci. 2018, 19, 3986. https://doi.org/10.3390/ijms19123986
Rodríguez-Vázquez L, Martí J. An Animal Model for Assessing the Effects of Hydroxyurea Exposure Suggests That the Administration of This Agent to Pregnant Women and Young Infants May Not Be as Safe as We Thought. International Journal of Molecular Sciences. 2018; 19(12):3986. https://doi.org/10.3390/ijms19123986
Chicago/Turabian StyleRodríguez-Vázquez, Lucía, and Joaquín Martí. 2018. "An Animal Model for Assessing the Effects of Hydroxyurea Exposure Suggests That the Administration of This Agent to Pregnant Women and Young Infants May Not Be as Safe as We Thought" International Journal of Molecular Sciences 19, no. 12: 3986. https://doi.org/10.3390/ijms19123986
APA StyleRodríguez-Vázquez, L., & Martí, J. (2018). An Animal Model for Assessing the Effects of Hydroxyurea Exposure Suggests That the Administration of This Agent to Pregnant Women and Young Infants May Not Be as Safe as We Thought. International Journal of Molecular Sciences, 19(12), 3986. https://doi.org/10.3390/ijms19123986