Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role




Abstract
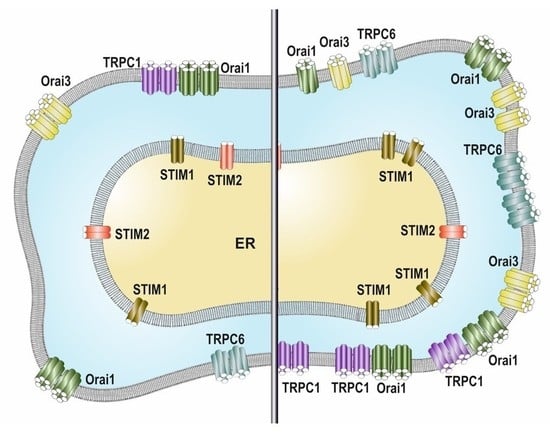
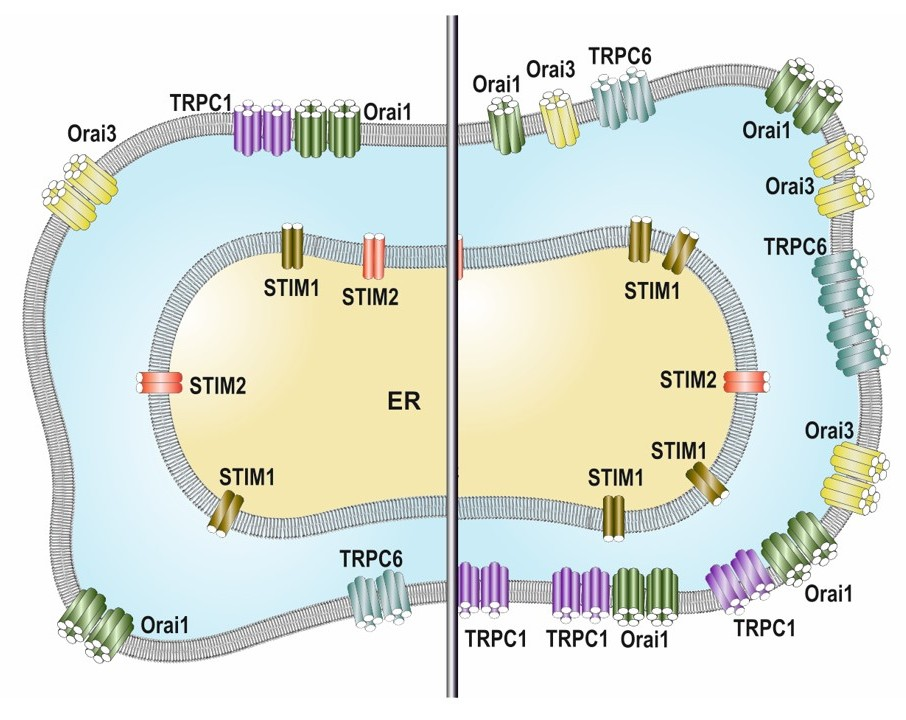
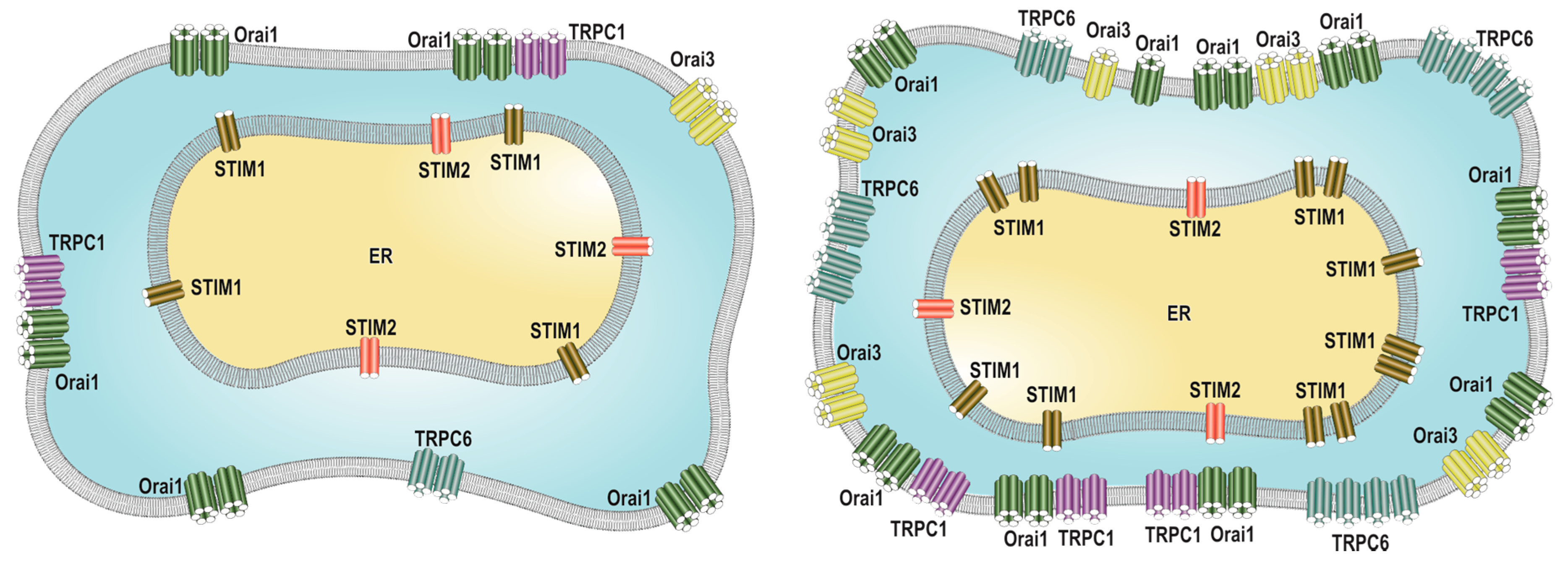
:
1. Molecular Basis of SOCE
1.1. STIM, Orai and TRPC Proteins
1.2. Activation of SOCE in Healthy Cells
2. SOCE Remodeling in Breast Cancer
2.1. SOCE in Breast Cancer
2.2. SOCE Remodeling in Breast Cancer Cells
3. Overview of Other Orai and TRPC-Dependent Ca2+ Influx in Breast Cancer Cells
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Putney, J.W. The physiological function of store-operated calcium entry. Neurochem. Res. 2011, 36, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Zheng, L.; Li, G.Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Zbidi, H.; Jardin, I.; Woodard, G.E.; Lopez, J.J.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A. STIM1 and STIM2 Are Located in the Acidic Ca2+ Stores and Associates with Orai1 upon Depletion of the Acidic Stores in Human Platelets. J. Biol. Chem. 2011, 286, 12257–12270. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell. Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Schindl, R.; Romanin, C. Studies of Structure-Function and Subunit Composition of Orai/STIM Channel. In Calcium Entry Channels in Non-Excitable Cells, 2018/10/10 ed.; Kozak, J.A., Putney, J.W., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2018; pp. 25–50. [Google Scholar]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Miederer, A.M.; Alansary, D.; Schwar, G.; Lee, P.H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjo, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Jardin, I.; Salido, G.M.; Rosado, J.A. Role of STIM2 in cell function and physiopathology. J. Physiol. 2017, 595, 3111–3128. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Madl, J.; Schutz, G.; Romanin, C. Structure, regulation and biophysics of I(CRAC), STIM/Orai1. Adv. Exp. Med. Biol. 2012, 740, 383–410. [Google Scholar] [PubMed]
- Muik, M.; Fahrner, M.; Derler, I.; Schindl, R.; Bergsmann, J.; Frischauf, I.; Groschner, K.; Romanin, C. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J. Biol. Chem. 2009, 284, 8421–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell. Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaoui, M.; Mimsy-Julienne, C.; Saby, C.; Van Gulick, L.; Peretti, M.; Jeannesson, P.; Morjani, H.; Ouadid-Ahidouch, H. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10.1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget 2018, 9, 24653–24671. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef]
- Scrimgeour, N.; Litjens, T.; Ma, L.; Barritt, G.J.; Rychkov, G.Y. Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J. Physiol. 2009, 587, 2903–2918. [Google Scholar] [CrossRef] [Green Version]
- Brechard, S.; Melchior, C.; Plancon, S.; Schenten, V.; Tschirhart, E.J. Store-operated Ca2+ channels formed by TRPC1, TRPC6 and Orai1 and non-store-operated channels formed by TRPC3 are involved in the regulation of NADPH oxidase in HL-60 granulocytes. Cell. Calcium 2008. [Google Scholar] [CrossRef]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J. Biol. Chem. 2008, 283, 25296–25304. [Google Scholar] [CrossRef]
- Kim, M.S.; Zeng, W.; Yuan, J.P.; Shin, D.M.; Worley, P.F.; Muallem, S. Native Store-operated Ca2+ Influx Requires the Channel Function of Orai1 and TRPC1. J. Biol. Chem. 2009, 284, 9733–9741. [Google Scholar] [CrossRef]
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Ong, E.C.; Nesin, V.; Long, C.L.; Bai, C.X.; Guz, J.L.; Ivanov, I.P.; Abramowitz, J.; Birnbaumer, L.; Humphrey, M.B.; Tsiokas, L. A TRPC1 protein-dependent pathway regulates osteoclast formation and function. J. Biol. Chem. 2013, 288, 22219–22232. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, T.J.; Thompson, J.L.; Mignen, O. STIM1 and the noncapacitative ARC channels. Cell. Calcium 2007, 42, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuttleworth, T.J. Arachidonic acid, ARC channels, and Orai proteins. Cell. Calcium 2009, 45, 602–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarran, L.; Lopez, J.J.; Woodard, G.E.; Salido, G.M.; Rosado, J.A. Store-operated Ca2+ entry-associated regulatory factor (SARAF) plays an important role in the regulation of arachidonate-regulated Ca2+ (ARC) channels. J. Biol. Chem. 2016, 291, 6982–6988. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.J.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. SARAF modulates TRPC1, but not TRPC6, channel function in a STIM1-independent manner. Biochem. J. 2016, 473, 3581–3595. [Google Scholar] [CrossRef]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef]
- Albarran, L.; Regodon, S.; Salido, G.M.; Lopez, J.J.; Rosado, J.A. Role of STIM1 in the surface expression of SARAF. Channels (Austin) 2016. [Google Scholar] [CrossRef]
- Jha, A.; Ahuja, M.; Maleth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell. Biol. 2013, 202, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarran, L.; Lopez, J.J.; Ben Amor, N.; Martín-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell. Sci. 2012, 125, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.W.; Trebak, M. Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal. 2015, 8, ra74. [Google Scholar] [CrossRef] [PubMed]
- Saotome, K.; Singh, A.K.; Yelshanskaya, M.V.; Sobolevsky, A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature 2016, 534, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP channels: An overview. Cell. Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Phelps, C.B.; Huang, R.J.; Lishko, P.V.; Wang, R.R.; Gaudet, R. Structural analyses of the ankyrin repeat domain of TRPV6 and related TRPV ion channels. Biochemistry 2008, 47, 2476–2484. [Google Scholar] [CrossRef]
- Rosado, J.A.; Sage, S.O. Activation of store-mediated calcium entry by secretion-like coupling between the inositol 1,4,5-trisphosphate receptor type II and human transient receptor potential (hTrp1) channels in human platelets. Biochem. J. 2001, 356, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; McGoldrick, L.L.; Twomey, E.C.; Sobolevsky, A.I. Mechanism of calmodulin inactivation of the calcium-selective TRP channel TRPV6. Sci. Adv. 2018, 4, eaau6088. [Google Scholar] [CrossRef]
- Dionisio, N.; Albarran, L.; Berna-Erro, A.; Hernandez-Cruz, J.M.; Salido, G.M.; Rosado, J.A. Functional role of the calmodulin- and inositol 1,4,5-trisphosphate receptor-binding (CIRB) site of TRPC6 in human platelet activation. Cell Signal. 2011, 23, 1850–1856. [Google Scholar] [CrossRef]
- Gregorio-Teruel, L.; Valente, P.; Gonzalez-Ros, J.M.; Fernandez-Ballester, G.; Ferrer-Montiel, A. Mutation of I696 and W697 in the TRP box of vanilloid receptor subtype I modulates allosteric channel activation. J. Gen. Physiol. 2014, 143, 361–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salido, G.M.; Jardin, I.; Rosado, J.A. The TRPC Ion Channels: Association with Orai1 and STIM1 Proteins and Participation in Capacitative and Non-capacitative Calcium Entry. Adv. Exp. Med. Biol. 2011, 704, 413–433. [Google Scholar] [PubMed]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat. Cell. Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Berna-Erro, A.; Smani, T.; Camello, P.J.; Salido, G.M.; Rosado, J.A. EFHB is a novel cytosolic Ca2+ sensor that modulates STIM1-SARAF interaction. Cell Physiol. Biochem. 2018, 51, 1164–1178. [Google Scholar]
- Cross, B.M.; Breitwieser, G.E.; Reinhardt, T.A.; Rao, R. Cellular calcium dynamics in lactation and breast cancer: From physiology to pathology. Am. J. Physiol. Cell Physiol. 2014, 306, 515–526. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Bolanz, K.A.; Hediger, M.A.; Landowski, C.P. The role of TRPV6 in breast carcinogenesis. Mol. Cancer Ther. 2008, 7, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Chodon, D.; Guilbert, A.; Dhennin-Duthille, I.; Gautier, M.; Telliez, M.S.; Sevestre, H.; Ouadid-Ahidouch, H. Estrogen regulation of TRPM8 expression in breast cancer cells. BMC Cancer 2010, 10, 212. [Google Scholar] [CrossRef]
- Diez-Bello, R.; Jardin, I.; Lopez, J.J.; El Haouari, M.; Ortega-Vidal, J.; Altarejos, J.; Salido, G.M.; Salido, S.; Rosado, J.A. (-)Oleocanthal inhibits proliferation and migration by modulating Ca2+ entry through TRPC6 in breast cancer cells. Biochim. Biophys. Acta Mol. Cell. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Grange, C.; Antoniotti, S.; Tomatis, C.; Merlino, A.; Bussolati, B.; Munaron, L. Arachidonic acid-induced Ca2+ entry is involved in early steps of tumor angiogenesis. Mol. Cancer Res. 2008, 6, 535–545. [Google Scholar] [PubMed]
- Yeh, Y.A.; Herenyiova, M.; Weber, G. Quercetin: synergistic action with carboxyamidotriazole in human breast carcinoma cells. Life Sci. 1995, 57, 1285–1292. [Google Scholar] [PubMed]
- Prasad, V.V.; Gopalan, R.O. Continued use of MDA-MB-435, a melanoma cell line, as a model for human breast cancer, even in year, 2014. NPJ Breast Cancer 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Oishi, Y.; Doi, I.; Shibata, H.; Kojima, I. Inhibition of proliferation of MCF-7 breast cancer cells by a blocker of Ca2+-permeable channel. Cell Calcium 1997, 22, 75–82. [Google Scholar] [CrossRef]
- Sergeev, I.N.; Rhoten, W.B. Regulation of intracellular calcium in human breast cancer cells. Endocrine 1998, 9, 321–327. [Google Scholar] [CrossRef]
- Greco, S.; Elia, M.G.; Muscella, A.; Storelli, C.; Marsigliante, S. AT1 angiotensin II receptor mediates intracellular calcium mobilization in normal and cancerous breast cells in primary culture. Cell Calcium 2002, 32, 1–10. [Google Scholar] [CrossRef]
- Rossi, A.M.; Picotto, G.; de Boland, A.R.; Boland, R.L. Evidence on the operation of ATP-induced capacitative calcium entry in breast cancer cells and its blockade by 17beta-estradiol. J. Cell Biochem. 2002, 87, 324–333. [Google Scholar] [CrossRef]
- Baldi, C.; Vazquez, G.; Boland, R. Capacitative calcium influx in human epithelial breast cancer and non-tumorigenic cells occurs through Ca2+ entry pathways with different permeabilities to divalent cations. J. Cell Biochem. 2003, 88, 1265–1272. [Google Scholar] [CrossRef]
- Gueder, N.; Allan, G.; Telliez, M.S.; Hague, F.; Fernandez, J.M.; Sanchez-Fernandez, E.M.; Ortiz-Mellet, C.; Ahidouch, A.; Ouadid-Ahidouch, H. sp(2) -Iminosugar alpha-glucosidase inhibitor 1-C-octyl-2-oxa-3-oxocastanospermine specifically affected breast cancer cell migration through Stim1, beta1-integrin, and FAK signaling pathways. J. Cell Physiol. 2017, 232, 3631–3640. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, X.; Shen, Q.; Yang, X.; Yu, C.; Cai, C.; Cai, G.; Meng, X.; Zou, F. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem. Biophys. Res. Commun. 2015, 458, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Mo, P.; Yang, S. The store-operated calcium channels in cancer metastasis: From cell migration, invasion to metastatic colonization. Front. Biosci. 2018, 23, 1241–1256. [Google Scholar]
- Cheng, H.; Wang, S.; Feng, R. STIM1 plays an important role in TGF-beta-induced suppression of breast cancer cell proliferation. Oncotarget 2016, 7, 16866–16878. [Google Scholar]
- Emeriau, N.; de Clippele, M.; Gailly, P.; Tajeddine, N. Store operated calcium entry is altered by the inhibition of receptors tyrosine kinase. Oncotarget 2018, 9, 16059–16073. [Google Scholar] [Green Version]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell Mol. Life Sci. 2018. [Google Scholar] [CrossRef]
- Ouwerkerk, R.; Jacobs, M.A.; Macura, K.J.; Wolff, A.C.; Stearns, V.; Mezban, S.D.; Khouri, N.F.; Bluemke, D.A.; Bottomley, P.A. Elevated tissue sodium concentration in malignant breast lesions detected with non-invasive 23Na MRI. Breast Cancer Res. Treat. 2007, 106, 151–160. [Google Scholar] [CrossRef]
- Amara, S.; Ivy, M.T.; Myles, E.L.; Tiriveedhi, V. Sodium channel gammaENaC mediates IL-17 synergized high salt induced inflammatory stress in breast cancer cells. Cell. Immunol. 2016, 302, 1–10. [Google Scholar] [CrossRef]
- Babaer, D.; Amara, S.; Ivy, M.; Zhao, Y.; Lammers, P.E.; Titze, J.M.; Tiriveedhi, V. High salt induces P-glycoprotein mediated treatment resistance in breast cancer cells through store operated calcium influx. Oncotarget 2018, 9, 25193–25205. [Google Scholar] [Green Version]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Tang, W.; Wang, Y.; Shen, Q.; Wang, B.; Cai, C.; Meng, X.; Zou, F. Downregulation of ACE2/Ang-(1-7)/Mas axis promotes breast cancer metastasis by enhancing store-operated calcium entry. Cancer Lett. 2016, 376, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Ghosh, S.; Banerjee, B.; Santra, A.; Adhikary, A.; Misra, A.K.; Sen, P.C. Phemindole, a Synthetic Di-indole Derivative Maneuvers the Store Operated Calcium Entry (SOCE) to Induce Potent Anti-Carcinogenic Activity in Human Triple Negative Breast Cancer Cells. Front. Pharmacol. 2016, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Miao, Y.; Zheng, X.; Gong, Y.; Zhang, J.; Zou, F.; Cai, C. STIM1 and STIM2 differently regulate endogenous Ca2+ entry and promote TGF-beta-induced EMT in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 488, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qin, K.; Zhang, Y.; Gong, J.; Li, N.; Lv, D.; Xiang, R.; Tan, X. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+ influx in breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 411, 786–791. [Google Scholar] [CrossRef]
- Schaar, A.; Sukumaran, P.; Sun, Y.; Dhasarathy, A.; Singh, B.B. TRPC1-STIM1 activation modulates transforming growth factor beta-induced epithelial-to-mesenchymal transition. Oncotarget 2016, 7, 80554–80567. [Google Scholar] [CrossRef]
- Pu, Q.; Zhao, Y.; Sun, Y.; Huang, T.; Lin, P.; Zhou, C.; Qin, S.; Singh, B.B.; Wu, M. TRPC1 intensifies house dust mite-induced airway remodeling by facilitating epithelial-to-mesenchymal transition and STAT3/NF-kappaB signaling. FASEB J. 2018. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Wang, Y.; Chen, Z.; Hua, D.; Yao, X.; Ma, X.; Zhang, P. Orai1 is critical for Notch-driven aggressiveness under hypoxic conditions in triple-negative breast cancers. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 975–986. [Google Scholar] [CrossRef]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 691–705. [Google Scholar] [CrossRef]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers (Basel) 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Kaemmerer, E.; Turner, D.; Peters, A.A.; Roberts-Thomson, S.J.; Monteith, G.R. An automated epifluorescence microscopy imaging assay for the identification of phospho-AKT level modulators in breast cancer cells. J. Pharmacol. Toxicol. Methods 2018, 92, 13–19. [Google Scholar] [CrossRef]
- Azimi, I.; Milevskiy, M.J.G.; Kaemmerer, E.; Turner, D.; Yapa, K.; Brown, M.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells. J. Cell Sci. 2017, 130, 2292–2305. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium. 2018, 74, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Redondo, P.C.; Salido, G.M.; Rosado, J.A. Phosphatidylinositol 4,5-bisphosphate enhances store-operated calcium entry through hTRPC6 channel in human platelets. Biochim. Biophys. Acta 2008, 1783, 84–97. [Google Scholar] [CrossRef]
- Jardin, I.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. Dynamic interaction of hTRPC6 with the Orai1/STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca2+ entry pathways. Biochem. J. 2009, 420, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta 2013, 1833, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Albarran, L.; Salido, G.M.; Lopez, J.J.; Sage, S.O.; Rosado, J.A. Fine-tuning of store-operated calcium entry by fast and slow Ca2+-dependent inactivation: Involvement of SARAF. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Launay, P.; Fleig, A.; Perraud, A.L.; Scharenberg, A.M.; Penner, R.; Kinet, J.P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef]
- Funabashi, K.; Ohya, S.; Yamamura, H.; Hatano, N.; Muraki, K.; Giles, W.; Imaizumi, Y. Accelerated Ca2+ entry by membrane hyperpolarization due to Ca2+-activated K+ channel activation in response to histamine in chondrocytes. Am. J. Physiol. Cell Physiol. 2010, 298, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Chantome, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca2+ channels: the complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Chantome, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Gueguinou, M.; Pages, J.C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [PubMed]
- Clarysse, L.; Gueguinou, M.; Potier-Cartereau, M.; Vandecasteele, G.; Bougnoux, P.; Chevalier, S.; Chantome, A.; Vandier, C. cAMP-PKA inhibition of SK3 channel reduced both Ca2+ entry and cancer cell migration by regulation of SK3-Orai1 complex. Pflugers Arch. 2014, 466, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Geerts, D.; Ay, A.S.; Potier-Cartereau, M.; Ahidouch, A.; Ouadid-Ahidouch, H. Functional cooperation between KCa3.1 and TRPC1 channels in human breast cancer: Role in cell proliferation and patient prognosis. Oncotarget 2016, 7, 36419–36435. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef]
- Dang, D.; Prasad, H.; Rao, R. Secretory pathway Ca2+-ATPases promote in vitro microcalcifications in breast cancer cells. Mol. Carcinog 2017, 56, 2474–2485. [Google Scholar] [CrossRef]
- Mignen, O.; Constantin, B.; Potier-Cartereau, M.; Penna, A.; Gautier, M.; Gueguinou, M.; Renaudineau, Y.; Shoji, K.F.; Felix, R.; Bayet, E.; et al. Constitutive calcium entry and cancer: updated views and insights. Eur. Biophys. J. 2017, 46, 395–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Haren, N.; Sevestre, H.; Ouadid-Ahidouch, H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am. J. Physiol. Cell Physiol. 2009, 297, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, W.; Zhang, H.; Zha, L.; Xue, Y.; Wu, X.; Zou, F. Requirement for store-operated calcium entry in sodium butyrate-induced apoptosis in human colon cancer cells. Biosci Rep. 2012, 32, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Ding, X.; Zhou, K.; Shi, Y.; Ge, R.; Ren, G.; Jin, Y.; Wang, Y. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int. J. Cancer 2009, 125, 2281–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Subtype | Cell Line | SOCE Constituents | References |
| Estrogen receptor positive | MCF7 | STIM1, STIM2, Orai3 and TRPC6 | [50,53,82,83,84] |
| Triple Negative/Basal | MDA-MB-231 | STIM1, Orai1 and TRPC6 | [53,82,83,84] |
| Breast Cancer Subtype | Cell Line | SOCE Remodeling | References |
| Estrogen receptor positive | MCF7 | ↓STIM1, ↔STIM2, ↑Orai3 and ↑TRPC6 | [50,53,82,83,84] |
| HCC 1500 | ↔STIM1 | [83] | |
| ZR751 | ↔STIM1 | [83] | |
| T47D | ↓STIM1 | [83] | |
| BT474 | ↓STIM1 | [83] | |
| HER2 | HCC1569 | ↑TRPC1 | [85] |
| Triple Negative/Basal | MDA-MB-231 | ↔STIM1, ↑Orai1, ↑TRPC1 and ↑TRPC6 | [53,82,83,84,86] |
| MDA-MB-468 | ↑TRPC1 | [86] | |
| BT20 | ↑STIM1 | [83] | |
| HCC 1937 | ↔STIM1 | [83] | |
| Patients | ↑STIM1, ↓STIM2 and ↑TRPC6 | [49] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. Int. J. Mol. Sci. 2018, 19, 4053. https://doi.org/10.3390/ijms19124053
Jardin I, Lopez JJ, Salido GM, Rosado JA. Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. International Journal of Molecular Sciences. 2018; 19(12):4053. https://doi.org/10.3390/ijms19124053
Chicago/Turabian StyleJardin, Isaac, Jose J. Lopez, Gines M. Salido, and Juan A. Rosado. 2018. "Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role" International Journal of Molecular Sciences 19, no. 12: 4053. https://doi.org/10.3390/ijms19124053
APA StyleJardin, I., Lopez, J. J., Salido, G. M., & Rosado, J. A. (2018). Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. International Journal of Molecular Sciences, 19(12), 4053. https://doi.org/10.3390/ijms19124053