Metabolic Signaling into Chromatin Modifications in the Regulation of Gene Expression


Abstract
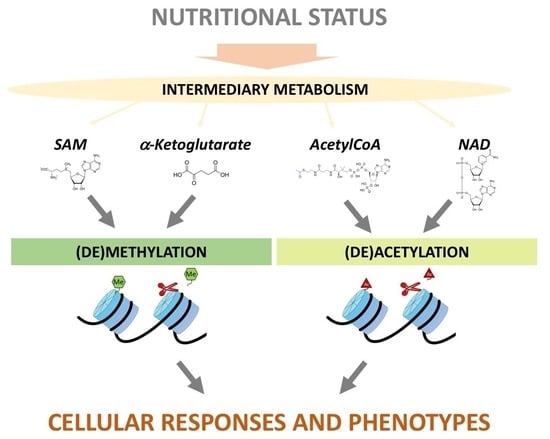
:
{kind=link}
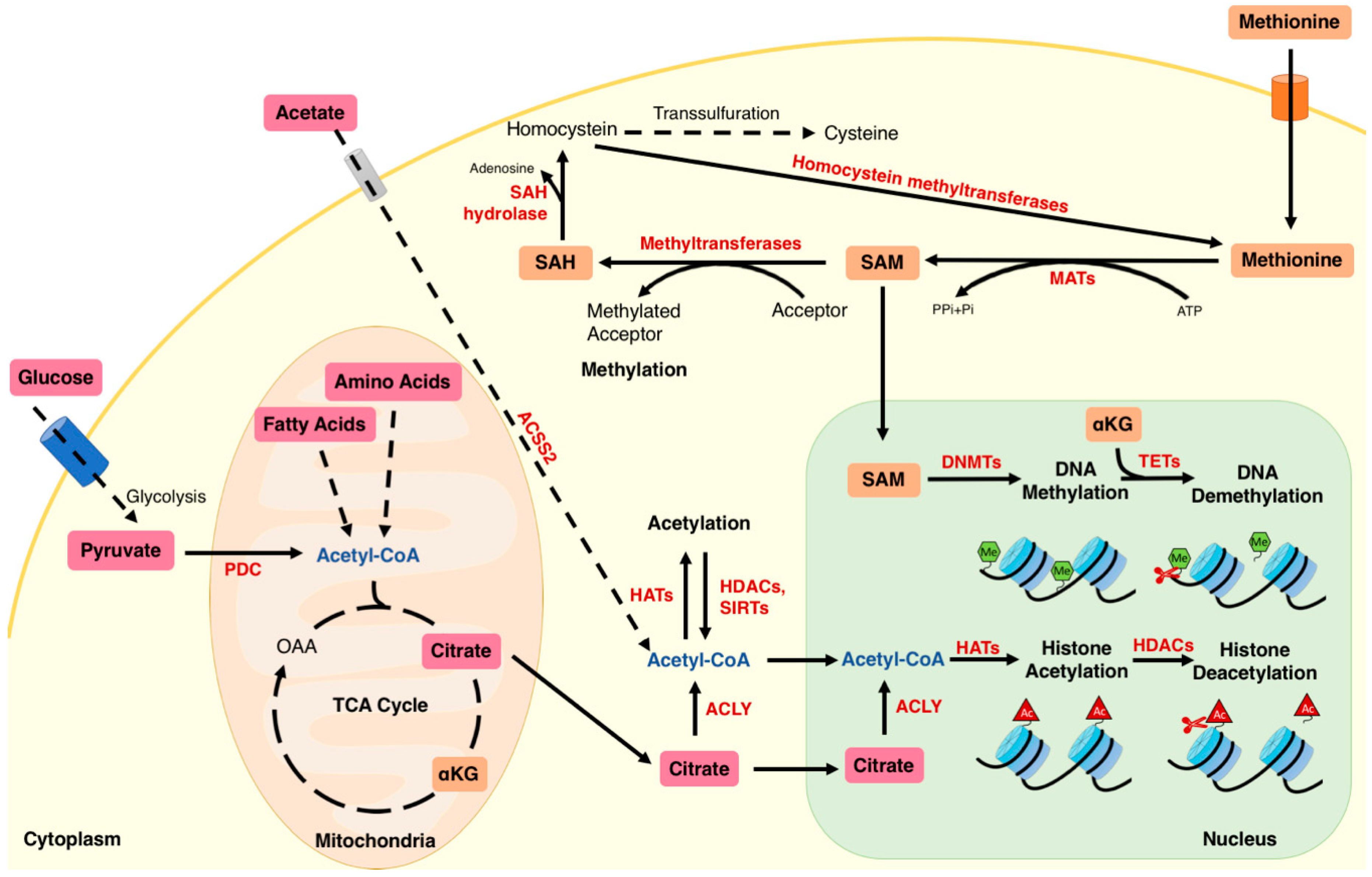{kind=link}
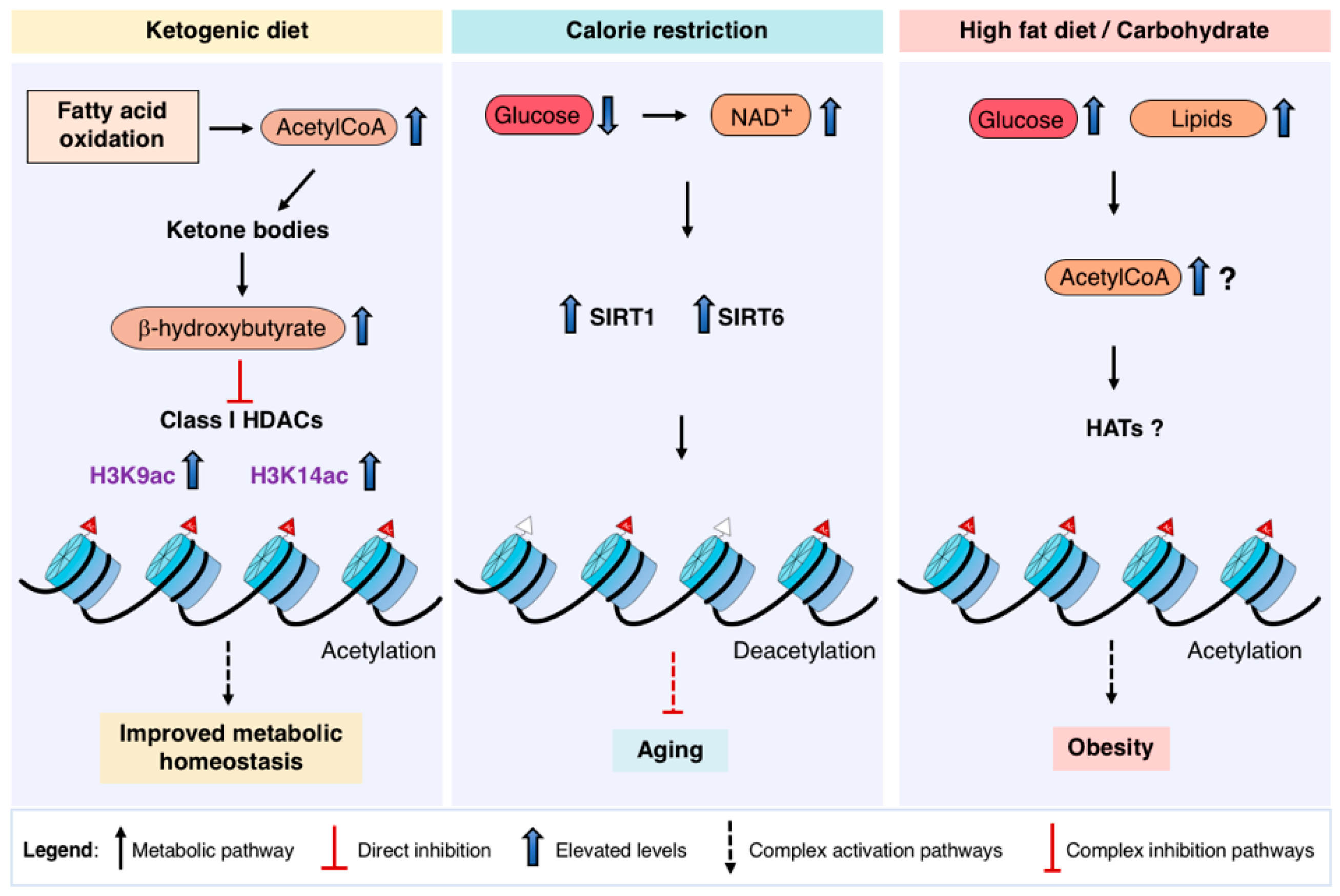{kind=link}
1. Introduction
2. Interaction of Metabolites with Chromatin Modifications
2.1. Acetyl-CoA and Regulation of Acetylation
2.2. NAD+ and Regulation of Sirtuin-Dependent Deacetylation
2.3. Methionine and Regulation of Methylation
2.4. α-Ketoglutarate (αKG) and Regulation of Demethylation
3. Metabolic-Chromatin Signaling in Different Physiological Contexts
3.1. Metabolic Reprogramming in Stem Cells
3.2. Metabolic Reprogramming in Immune Cells
4. Metabolic Signaling in the Regulation of Gene Expression of Metabolic Disorders
5. Therapeutic Diet Interventions Targeting Metabolic-Chromatin Axis
5.1. Ketogenesis
5.2. Calorie Restriction
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Manuel, J.; Pedro, B.-S.; Madeo, F.; Kroemer, G. Cell Metabolism Review Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA Induces Cell Growth and Proliferation by Promoting the Acetylation of Histones at Growth Genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena Histone Acetyltransferase A: A Homolog to Yeast Gcn5p Linking Histone Acetylation to Gene Activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.W.; Nandu, T.; Kim, J.; Challa, S.; DeBerardinis, R.J.; Kraus, W.L. Metabolic regulation of transcription through compartmentalized NAD+ biosynthesis. Science 2018, 360, eaan5780. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265.e6. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Zhang, H.; Katsyuba, E.; Auwerx, J. Protein acetylation in metabolism—Metabolites and cofactors. Nat. Rev. Endocrinol. 2016, 12, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Messner, S.; Hottiger, M.O. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011, 21, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Su, X.; Quinn, W.J.; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, J.T.; Brosnan, M.E.; Bertolo, R.; Brunton, J.A. Methionine: A metabolically unique amino acid. Livest. Sci. 2007, 112, 2–7. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Ueki, I. Dealing with methionine/homocysteine sulfur: Cysteine metabolism to taurine and inorganic sulfur. J. Inherit. Metab. Dis. 2011, 34, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14, R139–R147. [Google Scholar] [CrossRef] [PubMed]
- Comb, M.; Goodman, H.M. CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP-2. Nucleic Acids Res. 1990, 18, 3975–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Marion, D.W.; Cornatzer, W.E.; Duerre, J.A. S-Adenosylmethionine and S-adenosylhomocystein metabolism in isolated rat liver. Effects of L-methionine, L-homocystein, and adenosine. J. Biol. Chem. 1980, 255, 10822–10827. [Google Scholar] [PubMed]
- Shyh-Chang, N.; Locasale, J.W.; Lyssiotis, C.A.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.-F.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Surani, M.A.; Hajkova, P. Epigenetic Reprogramming of Mouse Germ Cells toward Totipotency. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, S.K.T.; Bestor, T.H.; Pfeifer, G.P. The colorful history of active DNA demethylation. Cell 2008, 133, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Zhang, Y. Reversing DNA Methylation: Mechanisms, Genomics, and Biological Functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-C.; Ng, H.-H.H. The metabolic programming of stem cells. Genes Dev. 2017, 31, 336–346. [Google Scholar] [CrossRef]
- Alexandre, G.-M.; Alajem, A.; Meshorer, E.; Miguel, R.-S. Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol. 2010, 12, 36–47. [Google Scholar] [CrossRef]
- Ying, Q.-L.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, D.C.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gómez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Mentch, S.J.; Gao, X.; Nichenametla, S.N.; Locasale, J.W. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat. Commun. 2018, 9, 1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Belmonte, J.C. Metabolic exit from naive pluripotency. Nat. Cell Biol. 2015, 17, 1519–1521. [Google Scholar] [CrossRef] [PubMed]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shai, S.-O.; Laevsky, I.; Amit, M.; et al. Glycolysis-Mediated Changes in Acetyl-CoA and Histone Acetylation Control the Early Differentiation of Embryonic Stem Cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryall, J.G.; Cliff, T.; Dalton, S.; Sartorelli, V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell 2015, 17, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Stefania, D.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gustavo, G.-C.; Fulco, M.; et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, K.; Chawla, A. Metabolic Regulation of Immune Responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hume, D.A. The Many Alternative Faces of Macrophage Activation. Front. Immunol. 2015, 6, 370. [Google Scholar] [CrossRef] [PubMed]
- Hard, G.C. Some biochemical aspects of the immune macrophage. Br. J. Exp. Pathol. 1970, 51, 97. [Google Scholar] [PubMed]
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 2012, 287, 25758–25769. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, G.; Apovian, C.M. Current pharmacotherapy for obesity. Nat. Rev. Endocrinol. 2018, 14, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Jenab, M.; Gunter, M.J. Adiposity and gastrointestinal cancers: Epidemiology, mechanisms and future directions. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, S.; Guénantin, A.-C.; Antonio, V.-P. ‘Basic and Applied Thermogenesis Research’ Bridging the Gap. Trends Endocrinol. Metab. 2017, 29, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sadhukhan, S.; Sun, S.; Wagner, G.R.; Hirschey, M.D.; Qi, L.; Lin, H.; Locasale, J.W. High-Resolution Metabolomics with Acyl-CoA Profiling Reveals Widespread Remodeling in Response to Diet. Mol. Cell. Proteom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Parris, J.L.D.; Trefely, S.; Henry, R.A.; Montgomery, D.C.; AnnMarie Torres, X.; Viola, J.M.; Kuo, Y.-M.; Blair, I.A.; Meier, J.L.; et al. Impact of a High-fat Diet on Tissue Acyl-CoA and Histone Acetylation Levels. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Eoin, M.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef]
- Inagaki, T.; Sakai, J.; Kajimura, S. Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nat. Rev. Mol. Cell Biol. 2016, 17, 480–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Liang, X.; Sun, X.; Zhang, L.; Fu, X.; Rogers, C.J.; Berim, A.; Zhang, S.; Wang, S.; Wang, B.; et al. AMPK/α-Ketoglutarate Axis Dynamically Mediates DNA Demethylation in the Prdm16 Promoter and Brown Adipogenesis. Cell Metab. 2016, 24, 542–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateishi, K.; Okada, Y.; Kallin, E.M.; Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009, 458, 757–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Rozqie, R.; Matsumura, Y.; Kawamura, T.; Nakaki, R.; Tsurutani, Y.; Tanimura-Inagaki, K.; Shiono, A.; Magoori, K.; Nakamura, K.; et al. JMJD1A is a signal-sensing scaffold that regulates acute chromatin dynamics via SWI/SNF association for thermogenesis. Nat. Commun. 2015, 6, 7052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Fujiwara, Y.; Takahashi, H.; Matsumura, Y.; Sawada, T.; Jiang, S.; Nakaki, R.; Uchida, A.; Nagao, N.; Naito, M.; et al. Histone demethylase JMJD1A coordinates acute and chronic adaptation to cold stress via thermogenic phospho-switch. Nat. Commun. 2018, 9, 1566. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Jedrychowski, M.P.; Chen, Y.; Serag, S.; Lavery, G.G.; Gygi, S.P.; Spiegelman, B.M. Lysine-specific demethylase 1 promotes brown adipose tissue thermogenesis via repressing glucocorticoid activation. Gene Dev. 2016, 32, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Schoonjans, K.; Auwerx, J. Histone Methyl Transferases and Demethylases; Can They Link Metabolism and Transcription? Cell Metab. 2010, 12, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominy, J.E.; Lee, Y.; Gerhart-Hines, Z.; Puigserver, P. Nutrient-dependent regulation of PGC-1α’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim. Biophys. Acta 2009, 1804, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Emmett, M.J.; Lim, H.W.; Jager, J.; Richter, H.J.; Adlanmerini, M.; Peed, L.C.; Briggs, E.R.; Steger, D.J.; Ma, T.; Sims, C.A.; et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature 2017, 546, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Boukouris, A.E.; Zervopoulos, S.D.; Michelakis, E.D. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem. Sci. 2016, 41, 712–730. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.J.; Hang, H.; Stephens, J.M. Pyruvate dehydrogenase complex (PDC) subunits moonlight as interaction partners of phosphorylated STAT5 in adipocytes and adipose tissue. J. Biol. Chem. 2017, 292, 19733–19742. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Deelen, J.; Slagboom, E.P. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Van der Klaauw, A.A.; Farooqi, I.S. The Hunger Genes: Pathways to Obesity. Cell 2015, 161, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.J.; Cooper, N.; Dunkley, A.; Warren, F.C.; Ara, R.; Abrams, K.; Davies, M.J.; Khunti, K.; Sutton, A. A systematic review and mixed treatment comparison of pharmacological interventions for the treatment of obesity. Obes. Rev. 2012, 13, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Kucharski, R.; Maleszka, J.; Foret, S.; Maleszka, R. Nutritional control of reproductive status in honeybees via DNA methylation. Science 2008, 319, 1827–1830. [Google Scholar] [CrossRef] [PubMed]
- Drummen, M.; Tischmann, L.; Blandine, G.-C.; Adam, T.; Margriet, W.-P. Dietary Protein and Energy Balance in Relation to Obesity and Co-morbidities. Front. Endocrinol. 2018, 9, 443. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Tognini, P.; Murakami, M.; Liu, Y.; Eckel-Mahan, K.L.; Newman, J.C.; Verdin, E.; Baldi, P.; Paolo, S.-C. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 2017, 26, 523–538.e5. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Bordone, L.; Guarente, L. Calorie restriction, SIRT1 and metabolism: Understanding longevity. Nat. Rev. Mol. Cell Biol. 2005, 6, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Calorie restriction and sirtuins revisited. Gene Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.A.; Philippe, C.-R.; et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S. NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; Davide, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, T.; Díaz-Hirashi, Z.; Verdeguer, F. Metabolic Signaling into Chromatin Modifications in the Regulation of Gene Expression. Int. J. Mol. Sci. 2018, 19, 4108. https://doi.org/10.3390/ijms19124108
Gao T, Díaz-Hirashi Z, Verdeguer F. Metabolic Signaling into Chromatin Modifications in the Regulation of Gene Expression. International Journal of Molecular Sciences. 2018; 19(12):4108. https://doi.org/10.3390/ijms19124108
Chicago/Turabian StyleGao, Tian, Zyanya Díaz-Hirashi, and Francisco Verdeguer. 2018. "Metabolic Signaling into Chromatin Modifications in the Regulation of Gene Expression" International Journal of Molecular Sciences 19, no. 12: 4108. https://doi.org/10.3390/ijms19124108
APA StyleGao, T., Díaz-Hirashi, Z., & Verdeguer, F. (2018). Metabolic Signaling into Chromatin Modifications in the Regulation of Gene Expression. International Journal of Molecular Sciences, 19(12), 4108. https://doi.org/10.3390/ijms19124108