Acute Limb Ischemia—Much More Than Just a Lack of Oxygen

 , ,
, ,
Abstract
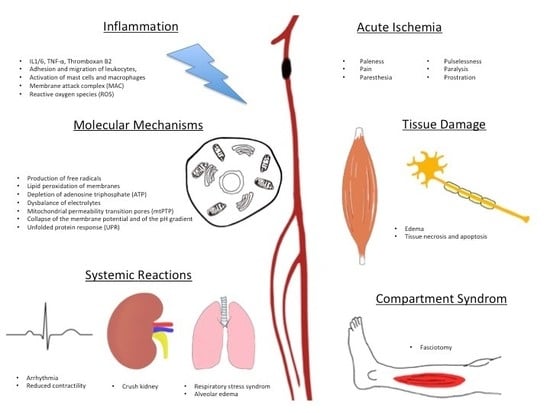
:
1. Introduction and Background
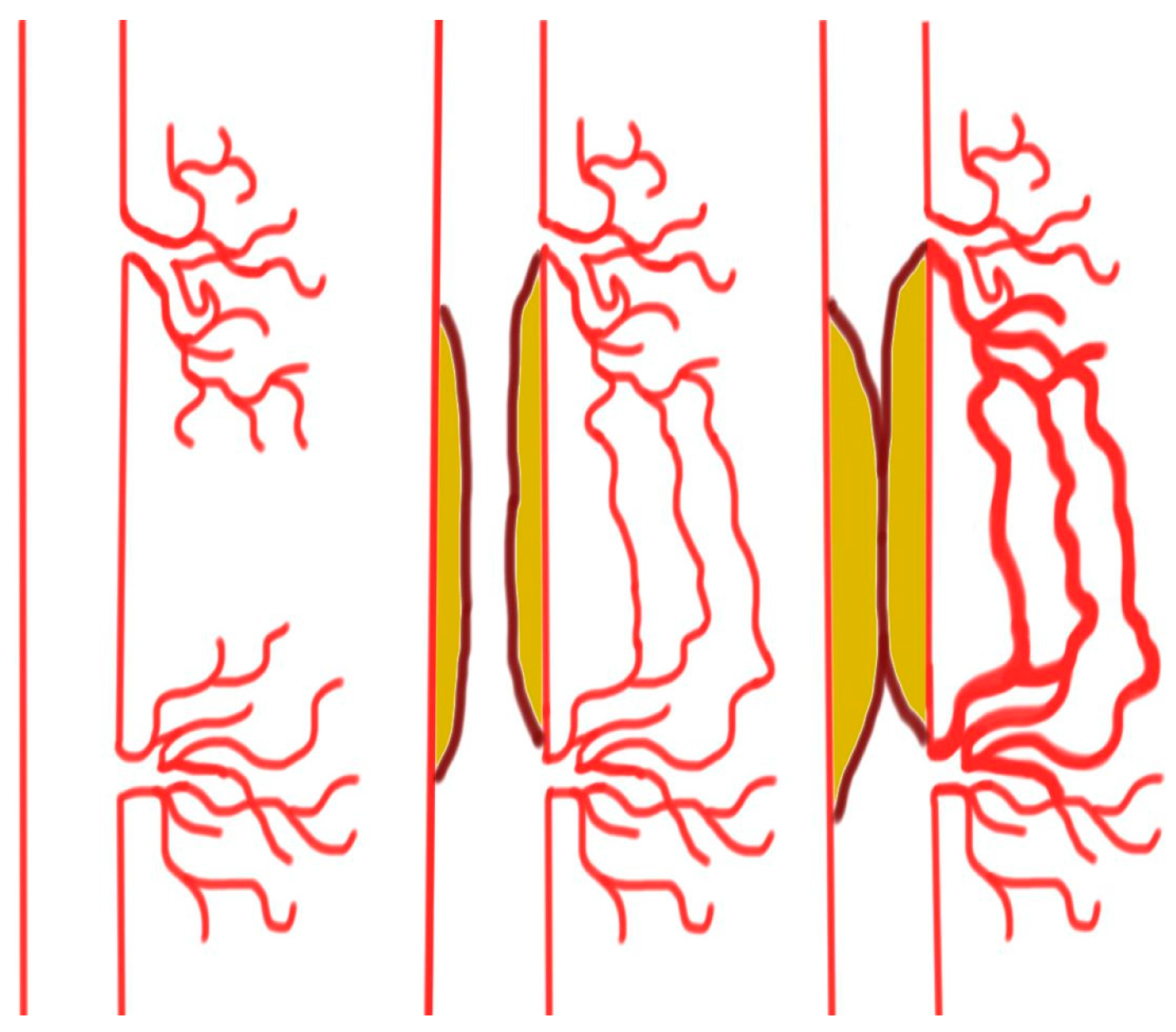
2. Collaterals
3. Edema
4. Inflammation
5. Molecular Mechanisms
6. Systemic Reactions
7. Treatment Options
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.; McDermott, M.; Norman, P.; Sampson, U.; Williams, L.; Mensah, G.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Lukasiewicz, A. Treatment of acute lower limb ischemia. Vasa 2016, 45, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Asakura, T.; Gotou, J.; Iwai, M.; Watanabe, Y.; Sando, M. Prediction of embolism in atrial fibrillation: Classification of left atrial thrombi by transesophageal echocardiography. Jpn. Circ. J. 2000, 64, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Clagett, G.P.; Sobel, M.; Jackson, M.R.; Lip, G.Y.; Tangelder, M.; Verhaeghe, R. Antithrombotic therapy in peripheral arterial occlusive disease: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004, 126, 609–626. [Google Scholar] [CrossRef] [PubMed]
- Menke, J.; Lüthje, L.; Kastrup, A.; Larsen, J. Thromboembolism in atrial fibrillation. Am. J. Cardiol. 2012, 105, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Saric, M.; Kronzon, I. Aortic atherosclerosis and embolic events. Curr. Cardiol. Rep. 2012, 14, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Buenger, C.M.; Knop, A.; Kische, S. Aortic dissection type B: Definition. Incidence, etiology. Gefässchirurgie 2015, 20, 415–419. [Google Scholar] [CrossRef]
- Rutherford, R.B.; Baker, J.D.; Ernst, C. Recommended standards for reports dealing with lower extremity ischemia: Revised version. J. Vasc. Surg. 1997, 26, 517–538. [Google Scholar] [CrossRef]
- Hummel, T.; Wenkel, M.; Papapostolou, G.; Mühlberger, D.; Mumme, A. Phlegmasia coerulea dolens. Diagnostics and therapy. Gefässchirurgie 2016, 21. [Google Scholar] [CrossRef]
- Koga, J.; Aikawa, M. Crosstalk between macrophages and smooth muscle cells in atherosclerotic vascular diseases. Vascul. Pharmacol. 2013, 57, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R.; Purushothaman, M.; Purushothaman, K.R. Plaque neovascularization: Defense mechanisms, betrayal, or a war in progress. Ann. N. Y. Acad. Sci. 2012, 1254, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Farb, A.; Malcom, G.T.; Liang, Y.; Smialek, J.E.; Virmani, R. Plaque rupture and sudden death related to exertion in men with coronary artery disease. JAMA 1999, 281, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Tedgui, A.; Mallat, Z. Apoptosis as a determinant of atherothrombosis. Thromb. Haemost. 2001, 86, 420–426. [Google Scholar] [PubMed]
- Tang, G.L.; Chang, D.S.; Sarkar, R.; Wang, R.; Messina, L.M. The effect of gradual or acute arterial occlusion on skeletal muscle blood flow, arteriogenesis, and inflammation in rat hindlimb ischemia. J. Vasc. Surg. 2005, 41, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Schaper, W. Collateral circulation: Past and present. Basic Res. Cardiol. 2009, 104, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Ito, W.; Fleming, I.; Deindl, E.; Sauer, A.; Wienet, M.; Busse, R.; Schaper, J.; Schaper, W. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Arch. 2000, 436, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Pratt, G.H.; Krahl, E. Surgical therapy for the occluded artery. Am. J. Surg. 1954, 87, 722–729. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.O.; Thal, E.R.; Shires, G.T. Management of arterial injuries. Ann. Surg. 1971, 173, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, M.H.; Oltmann, C.L.; Bowles, D.K. Exercise training-induced adaptations in the coronary circulation. Med. Sci. Sports Exerc. 1998, 30, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Sarelius, I.; Pohl, U. Control of muscle blood flow during exercise: Local factors and integrative mechanisms. Acta Physiol. 2010, 199, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, T.T.; Korpisalo, P.; Markkanen, J.E.; Liimatainen, T.; Ordén, M.R.; Kholová, I.; de Goede, A.; Heikura, T.; Gröhn, O.H.; Ylä-Herttuala, S. Blood flow remodels growing vasculature during vascular endothelial growth factor gene therapy and determines between capillary arterialization and sprouting angiogenesis. Circulation 2005, 112, 3937–3946. [Google Scholar] [CrossRef] [PubMed]
- Makanya, A.N.; Hlushchuk, R.; Djonov, V.G. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis 2009, 12, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF-System. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Dragneva, G.; Korpisalo, P.; Yla-Herttuala, S. Promoting blood vessel growth in ischemic diseases: Challenges in translating preclinical potential into clinical success. Dis. Model. Mech. 2013, 6, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular ednothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, U.R. Mechanisms of endothelial cell migration. Cell. Mol. Life Sci. 2014, 71, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Cai, W.J.; Schaper, W. Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis 2001, 4, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Simons, M. Angiogenesis: Where do we stand now? Circulation 2005, 111, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Egginton, S.; Hudlick, O. Selective long-term electrical stimulation of fast glycocalic fibres increases capillary supply but not oxidative enzyme activity in rat skeletal muscles. Exp. Physiol. 2000, 85, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, P.G.; Prior, B.M.; Yang, H.T.; Terjung, R.L. Angiogenic growth factor expression in rat skeletal muscle in response to exercise training. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, 1668–1678. [Google Scholar] [CrossRef] [PubMed]
- Schaper, W.; DeBrabander, M.; Lewi, P. DNA-synthesis and mitoses in coronary collateral vessels of the dog. Circ. Res. 1971, 28, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Van Royen, N.; Piek, J.J.; Buschmann, I.; Hoefer, I.; Voskuil, M.; Schaper, W. Stimulation of arteriogenesis; a new concept for the treatment of arterial occlusive disease. Cardiovasc. Res. 2001, 49, 543–553. [Google Scholar] [CrossRef]
- Saleh, F.H.; Jurjus, A.R. A comparative study of morphological changes in spontaneously hypertensive rats and normotensive Wistar Kyoto rats treated with an angiotensin-converting enzyme inhibitor or a calcium-channel blocker. J. Pathol. 2006, 193, 415–420. [Google Scholar] [CrossRef]
- Mullick, S. The tourniquet in operations upon the extremities. Surg. Gynecol. Obstet. 1978, 146, 821. [Google Scholar] [PubMed]
- Arató, E.; Kürthy, M.; Sínay, L.; Kasza, G.; Menyhei, G.; Masoud, S.; Bertalan, A.; Verzár, Z.; Kollár, L.; Roth, E.; et al. Pathology and diagnostic options of lower limb compartment syndrome. Clin. Hemorheol. Microcirc. 2009, 41, 1–8. [Google Scholar] [PubMed]
- Solonen, K.A.; Hjelt, L. Morphological changes in striated muscles during ischemia. Acta Orthop. Scand. 1968, 39, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Malan, E.; Tattoni, G. Physio- and anatomo-pathology of acute ischemia of the extremities. J. Cardiovasc. Surg. 1963, 4, 212–225. [Google Scholar]
- Matsen, F.; Winquist, R.; Krugmire, R. Diagnosis and management of compartmental syndromes. J. Bone Jt. Surg. 1980, 62, 286–291. [Google Scholar] [CrossRef]
- Fukuda, I.; Chiyoya, M.; Taniquchi, S.; Fukuda, W. Acute limb ischemia: Contemporary approach. Gen. Thorac. Cardiovasc. Surg. 2015, 63, 540. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Rehm, M.; Bruegger, D.; Christ, F.; Conzen, P.; Thiel, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Reichart, B.; et al. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation 2007, 116, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Dunant, J.H.; Edwards, W.S. Small vessel occlusion in the extremity after various periods of arterial obstruction: An experimental study. Surgery 1973, 73, 240–245. [Google Scholar] [PubMed]
- Massoudy, P.; Zahler, S.; Freyholdt, T.; Henze, R.; Barankay, A.; Becker, B.F.; Braun, S.L.; Meisner, H. Sodium nitroprusside in patients with compromised left ventricular function undergoing coronary bypass: Reduction of cardiac proinflammatory substances. J. Thorac. Cardiovasc. Surg. 2000, 119, 566–574. [Google Scholar] [CrossRef]
- Spencer, R.C.; Eiseman, B. Delayed arterial embolectomy. A new concept. Surgery 1964, 55, 64–72. [Google Scholar] [PubMed]
- Witthaut, R.; Farhood, A.; Smith, C.W.; Jaeschke, H. Complement and tumor necrosis factor-α contribute to Mac-1 (CD11b/CD18) up-regulation and systemic neutrophil activation during endotoxemia in vivo. J. Leukoc. Biol. 1994, 55, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S. Reactive oxygen species-induced molecular damage and its application in pathology. Pathol. Int. 1999, 49, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.G.; Gallin, J.I. A functional differentiation of human neutrophil granules: Generation of C5a by a specific (secondary) granule product and inactivation of C5a by azurophil (primary) granule products. J. Immunol. 1977, 119, 1068–1076. [Google Scholar] [PubMed]
- Oseas, R.; Yang, H.H.; Baehner, R.L.; Boxer, L.A. Lactoferrin: A promoter of polymorphonuclear leukocyte adhesiveness. Blood 1981, 57, 939–945. [Google Scholar] [PubMed]
- Kjeldsen, L.; Sengelov, H.; Lollike, K. Isolation and characterization of gelatinase granules from human neutrophils. Blood 1994, 83, 1640–1649. [Google Scholar] [PubMed]
- Weiss, S.J. Tissue destruction by neutrophils. N. Engl. J. Med. 1989, 320, 365–376. [Google Scholar] [PubMed]
- Vouldoukis, I.; Conti, M.; Krauss, P.; Kamaté, C.; Blazquez, S.; Tefit, M.; Mazier, D.; Calenda, A.; Dugas, B. Supplementation with gliadin-combined plant superoxide dismutase extract promotes antioxidant defences and protects against oxidative stress. Phytother. Res. 2004, 18, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Casey, D.P.; Joyner, M.J. Local control of skeletal muscle blood flow during exercise: Influence of available oxygen. J. Appl. Physiol. 2001, 111, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Green, C.J.; Hellberg, O. Evidence of free-radical-induced damage in rabbit kidneys after hypothermic preservation and autotransplantation. Transplantation 1986, 41, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Gelman, S. The pathophysiology of aortic cross-clamping and unclamping. Anesthesiology 1995, 82, 1026–1060. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood 1998, 92, 3007–3017. [Google Scholar] [PubMed]
- Smith, J.K.; Carden, D.L.; Korthuis, R.J. Role of xanthine oxidase in postischemic microvascular injury in skeletal muscle. Am. J. Physiol. 1989, 257, 1782–1789. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J. Properties, functions, and secretion of human myeloperoxidase. Biochemistry 2004, 6, 4–9. [Google Scholar] [CrossRef]
- Ozmen, S.; Ayhan, S.; Demir, Y.; Siemionow, M.; Atabay, K. Impact of gradual blood flow increase on ischaemia-reperfusion injury in the rat cremaster microcirculation model. J. Plast. Reconstr. Aesthet. Surg. 2008, 61, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Carnevale, R.; Cangemi, R.; Angelico, F.; Augelletti, T.; di Santo, S.; Calabrese, C.M.; della Volpe, L.; Pignatelli, P.; Perri, L. NOX2 up-regulation is associated with artery dysfunction in patients with peripheral artery disease. Int. J. Cardiol. 2013, 165, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.W.C.; Halestrap, A.P. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A. A pore way to die. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Halestrap, A.P. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem. Soc. Trans. 2006, 34, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; BlalchyDyson, E.; DiLisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Kroemer, G. Apoptosis: Mitochondrial membrane permeabilization—The (W)hole story. Curr. Biol. 2003, 13, 71–73. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Kroemer, G. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Grall, S.; Prunier-Mirebeau, D.; Tamareille, S.; Mateus, V.; Lamon, D.; Furber, A. Endoplasmic reticulum stress pathway involvement in local and remote myocardial ischemic conditioning. Shock 2013, 39, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Qin, J.; Qian, X.; Lu, L.; Wang, P.; Wu, Z. Lipopolysaccharide preconditioning protects hepatocytes from ischemia/reperfusion injury (IRI) through inhibiting ATF4-CHOP pathway in mice. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Degracia, D.J.; Montie, H.L. Cerebral ischemia and the unfolded protein response. J. Neurochem. 2004, 91, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Lee, A.S. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucl. Acids Res. 1999, 27, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 27, 13935–13940. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Haimovici, H. Metabolic complications of acute arterial occlusions. J. Cardiovasc. Surg. 1979, 20, 349–357. [Google Scholar]
- Carden, D.; Xiao, F.; Moak, C.; Willis, B.H.; Robinson-Jackson, S.; Alexander, S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am. J. Physiol. 1998, 275, 385–392. [Google Scholar] [CrossRef]
- Husmann, M.J.; Barton, M.; Amann-Vesti, B.R.; Franzeck, U.K. Postural effects on interstitial fluid pressure in humans. J. Vasc. Res. 2006, 43, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Alardin, A.L.; Varon, J.; Marik, P.E. Bench-to-bedside review: Rhabdomyolysis—An overview for clinicians. Crit. Care 2005, 9, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Welbourn, R.; Goldman, G.; O’Riordain, M. Role for tumor necrosis factor as mediator of lung injury following lower torso ischemia. J. Appl. Physiol. 1991, 70, 2645–2649. [Google Scholar] [CrossRef] [PubMed]
- Welbourn, R.; Goldman, G.; Kobzik, L. Role of neutrophil adherence receptors (CD 18) in lung permeability following lower torso ischemia. Circ. Res. 1992, 71, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Kyriakides, C.; Austen, W.G.; Wang, Y.; Favuzza, J.; Moore, F.D.; Hechtman, H.B. Neutrophil mediated remote organ injury after lower torso ischemia and reperfusion is selectin and complement dependent. J. Trauma 2000, 48, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Suratt, B.T.; Parsons, P.E. Mechanisms of acute lung injury/acute respiratory distress syndrome. Clin. Chest Med. 2006, 27, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.D. Miller’s Anesthesia, 6th ed.; Churchill Livingstone: New York, NY, USA, 2005. [Google Scholar]
- Marsh, J.D.; Margolis, T.I.; Kim, D. Mechanism of diminished contractile response to catecholamines during acidosis. Am. J. Physiol. 1988, 254, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Walley, K.R.; Hebert, P.C.; Wakai, Y.; Wilcox, P.G.; Road, J.D.; Cooper, D.J. Decrease in left ventricular contractility after tumor necrosis factor α infusion in dogs. J. Appl. Physiol. 1994, 76, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Samaja, M.; Allibardi, S.; Milano, G. Differential depression of myocardial function and metabolism by lactate and H+. Am. J. Physiol. 1999, 276, 3–8. [Google Scholar] [CrossRef]
- Lipsitz, E.C.; Veith, F.J. Fluoroscopically assisted thromboembolectomy: Should it be routine? Semin. Vasc. Surg. 2001, 14, 100–106. [Google Scholar] [CrossRef] [PubMed]
- De Donato, G.; Setacci, F.; Sirignano, P. The combination of surgical embolectomy and endovascular techniques may improve outcomes of patients with acute lower limb ischemia. J. Vasc. Surg. 2014, 59, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, M.; Varty, K.; Nydahl, S. The surgical management of acute limb ischaemia due to native vessel occlusion. Eur. J. Vasc. Endovasc. Surg. 1999, 17, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Comerota, A.J.; Sidhu, R. Can intraoperative thrombolytic therapy assist with the management of acute limb ischemia? Semin. Vasc. Surg. 2009, 22, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.O.; Berridge, D.C.; Robertson, I. Infusion techniques for peripheral arterial thrombolysis. Cochrane Database Syst. Rev. 2004. [Google Scholar] [CrossRef]
- Sniderman, K.W.; Bodner, L.; Saddekni, S. Percutaneous embolectomy by transcatheter aspiration. Work in progress. Radiology 1984, 150, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Canova, C.R.; Schneider, E.; Fischer, L. Long-term results of percutaneous thromboembolectomy in patients with infranguinal embolic occlusions. Int. Angiol. 2001, 20, 66–73. [Google Scholar] [PubMed]
- Zeller, T.; Frank, U.; Bürgelin, K. Early experience with a rotational thrombectomy device for treatment of acute and subacute infra-aortic arterial occlusions. J. Endovasc. Ther. 2003, 10, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberg, M.; Kunicke, M.; Hailer, B. Percutaneous mechanical thrombectomy for treatment of acute femoropopliteal bypass occlusion. Vasc. Health Risk. Manag. 2012, 8, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Kasirajan, K.; Gray, B.; Beavers, F.P. Rheolytic thrombectomy in the management of acute and subacute limb-threatening ischemia. J. Vasc. Interv. Radiol. 2001, 12, 413–421. [Google Scholar] [CrossRef]
- Barbato, J.E.; Wholey, M.H. Use of AngioJet mechanical thrombectomy for acute peripheral ischemia associated with stent fracture. Catheter. Cardiovasc. Interv. 2007, 70, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Borgia, F.; Di Serafino, L.; Sannino, A. AngioJet rheolytic thrombectomy for acute superficial femoral artery stent or femoropopliteal by-pass thrombosis. Monaldi Arch. Chest Dis. 2010, 74, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Spiliopoulos, S.; Katsanos, K.; Fragkos, G. Treatment of infrainguinal thromboembolic complications during peripheral endovascular procedures with AngioJet rheolytic thrombectomy, intraoperative thrombolysis, and selective stenting. J. Vasc. Surg. 2012, 56, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Ansel, G.M.; George, B.S.; Botti, C.F. Rheolytic thrombectomy in the management of limb ischemia: 30-day results from a multicenter registry. J. Endovasc. Ther. 2002, 9, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Piercy, K.T.; Ayerdi, J.; Geary, R.L. Acute pancreatitis: A complication associated with rheolytic mechanical thrombectomy of deep venous thrombosis. J. Vasc. Surg. 2006, 44, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Carrera, L.A.; Reddy, R.; Pamoukian, V.N. Massive intravascular hemolysis with mechanical rheolytic thrombectomy of a hemodialysis arteriovenous fistula. Semin. Dial. 2013, 26, 5–7. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6 Ps | Symptom | Explanation |
|---|---|---|
| 1. | Paleness | Paleness of the Skin |
| 2. | Pain | Aggravating ischemic pain |
| 3. | Paresthesia | Ascending paresthesia of the skin |
| 4. | Pulselessness | Absence of pulse |
| 5. | Paralysis | Ascending paralysis of the muscles |
| 6. | Prostration | Fatigue, shock, agitation |
| Class | Category | Prognosis | Sensory Loss | Muscle Weakness | Arterial Doppler | Venous Doppler |
|---|---|---|---|---|---|---|
| I | Viable | No immediate limb threat | None | None | Audible | Audible |
| IIA | Threatening:marginal | Salvageable if treated promptly | Minimal–none | None | Inaudible | Audible |
| IIB | Threatening:immediate | Salvageable if treated immediately | More than just toes | Mild–moderate | Inaudible | Audible |
| III | Irreversible | Limb loss or permanent damage | Profound | Profound | Inaudible | Inaudible |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, F.; Oberhuber, A.; Floros, N.; Busch, A.; Wagenhäuser, M.U.; Schelzig, H.; Duran, M. Acute Limb Ischemia—Much More Than Just a Lack of Oxygen. Int. J. Mol. Sci. 2018, 19, 374. https://doi.org/10.3390/ijms19020374
Simon F, Oberhuber A, Floros N, Busch A, Wagenhäuser MU, Schelzig H, Duran M. Acute Limb Ischemia—Much More Than Just a Lack of Oxygen. International Journal of Molecular Sciences. 2018; 19(2):374. https://doi.org/10.3390/ijms19020374
Chicago/Turabian StyleSimon, Florian, Alexander Oberhuber, Nikolaos Floros, Albert Busch, Markus Udo Wagenhäuser, Hubert Schelzig, and Mansur Duran. 2018. "Acute Limb Ischemia—Much More Than Just a Lack of Oxygen" International Journal of Molecular Sciences 19, no. 2: 374. https://doi.org/10.3390/ijms19020374
APA StyleSimon, F., Oberhuber, A., Floros, N., Busch, A., Wagenhäuser, M. U., Schelzig, H., & Duran, M. (2018). Acute Limb Ischemia—Much More Than Just a Lack of Oxygen. International Journal of Molecular Sciences, 19(2), 374. https://doi.org/10.3390/ijms19020374