The Many Facets of Metzincins and Their Endogenous Inhibitors: Perspectives on Ovarian Cancer Progression


Abstract
:
1. What Are Metzincins?
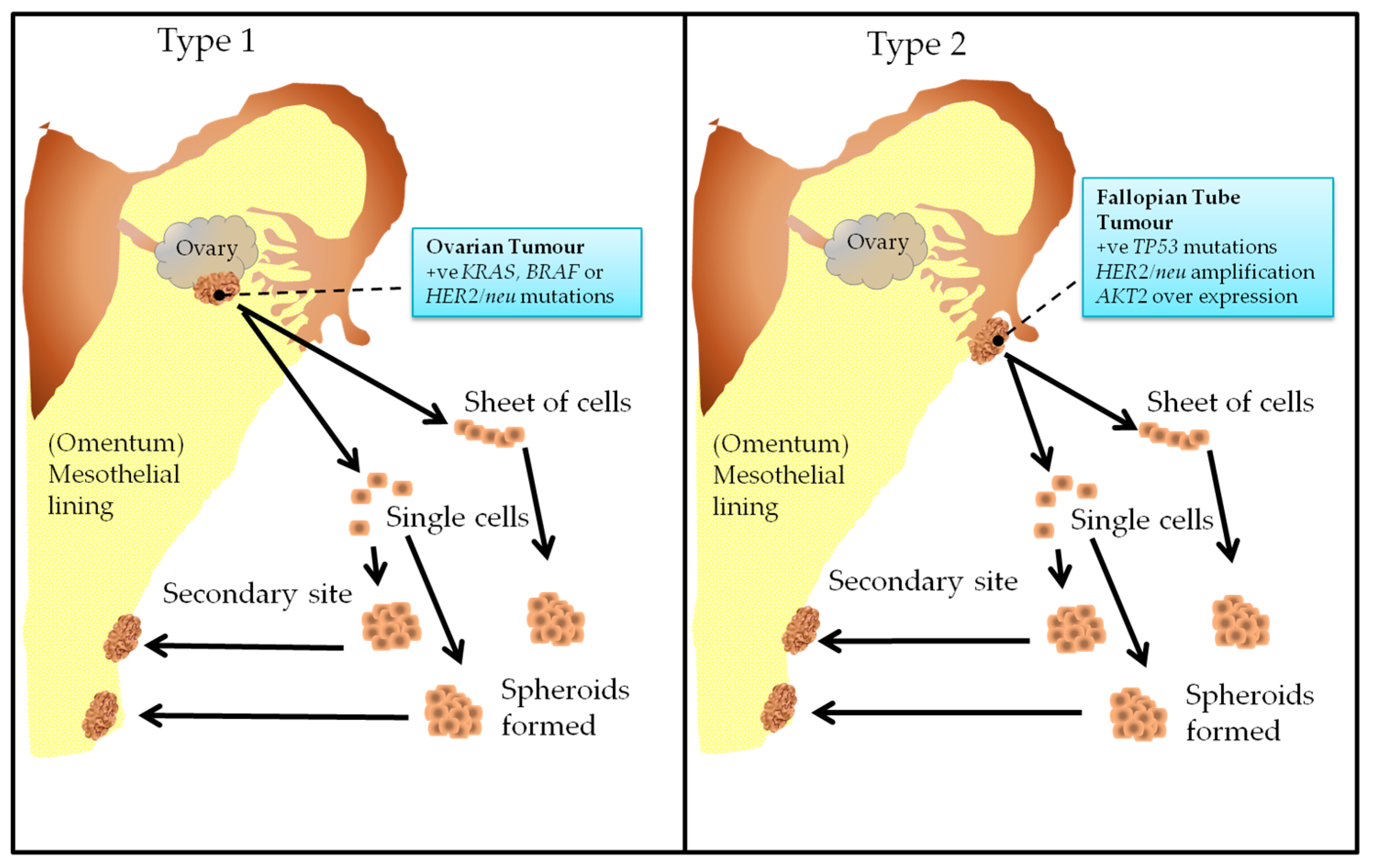
2. The Origin of Epithelial Ovarian Cancer
3. Recent Classification of Epithelial Ovarian Cancer
4. What Is Known about Ovarian Cancer Progression?
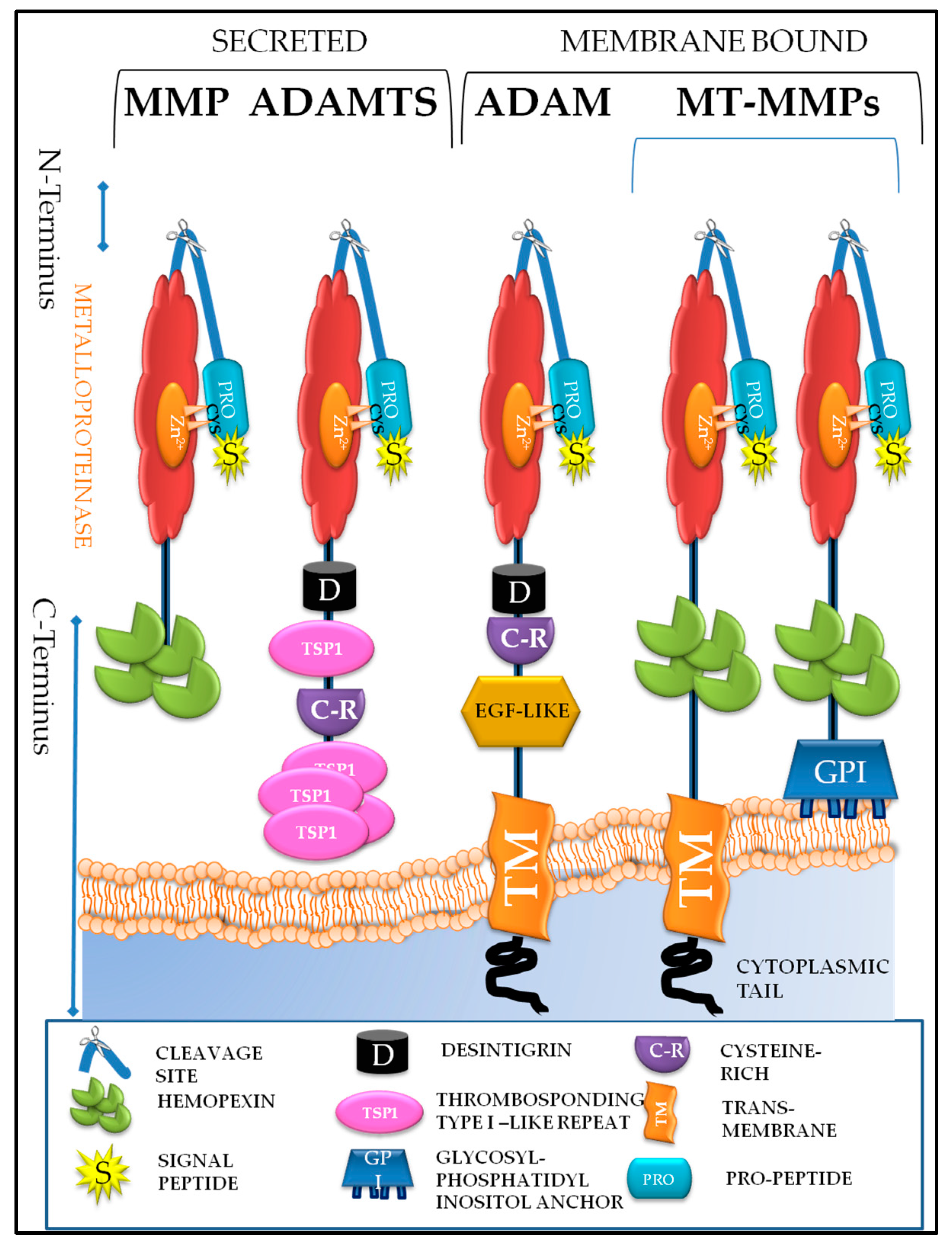
5. Understanding the Structure and Roles of Metzincins and Their Inhibitors
5.1. Matrixin Family of Metzincins
Matrix Metalloproteinases (MMPs)
5.2. Adamalysin Family of Metzincins
5.2.1. A Disintegrin and A Metalloproteinases (ADAMs)
5.2.2. ADAMS with Thrombospondin Motifs (ADAMTS)
6. What Are Metzincin Inhibitors?
6.1. Synthetic Inhibitors
6.2. Endogenous Inhibitors
6.3. Do TIMPs Only Inhibit Metzincin’s Activity or Are They Independent of Metzincin?
6.4. What Is the Expression of TIMPs in Tumors?
7. Metzincins and TIMPS in Ovarian Cancer
7.1. What Is the Evidence of the Involvement of MMPs and TIMPs in Ovarian Cancer?
7.1.1. Expression in Tumors, Ascites and Blood
7.1.2. Expression and Function in Cell Lines
7.2. Adamalysin in Ovarian Cancer
7.3. What Is the Role of Metzincins in Ovarian Cancer? and What Are the Underlying Mechanisms?
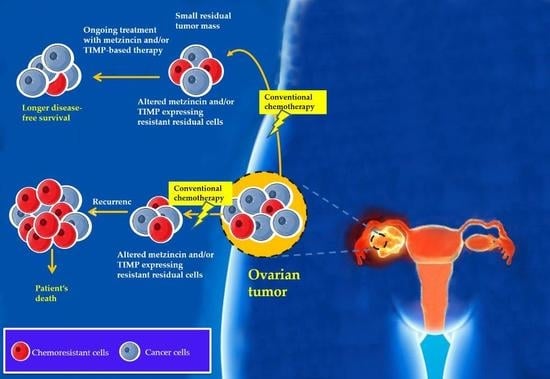
8. Chemotherapy Treatment Results in the Enhanced Expression of TIMP-1, -2 and -3 In Vitro and In Vivo: A Proof of Concept Experimental Model
9. Potential New Therapeutic Approaches for TIMPs
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Balaban, N.P.; Rudakova, N.L.; Sharipova, M.R. Structural and functional characteristics and properties of metzincins. Biochemistry 2012, 2, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Rivera, S.; Khrestchatisky, M.; Kaczmarek, L.; Rosenberg, G.A.; Jaworski, D.M. Metzincin Proteases and their Inhibitors, Foes or Friends in Nervous System Physiology? J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 15337–15357. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Chen, X.; Greenawalt, E.J.; Maulik, U.; Jiang, W.; Zhao, Z.; Eischen, C.M. Decoding critical long non-coding RNA in ovarian cancer epithelial-to-mesenchymal transition. Nat. Commun. 2017, 8, 1604. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Thompson, E.W.; Quinn, M.A. Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: An exception to the norm. J. Cell. Physiol. 2007, 213, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Wong, A.S.; Choi, K.C.; Kang, S.K.; Leung, P.C. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Levanon, K.; Crum, C.; Drapkin, R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.W.; Miron, A.; Jarboe, E.A.; Parast, M.M.; Hirsch, M.S.; Lee, Y.; Muto, M.G.; Kindelberger, D.; Crum, C.P. Serous tubal intraepithelial carcinoma: Its potential role in primary peritoneal serous carcinoma and serous cancer prevention. J. Clin. Oncol. 2008, 26, 4160–4165. [Google Scholar] [CrossRef] [PubMed]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Kim, O.; Stack, M.S. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers 2017, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Vang, R.; Shih, I.M.; Kurman, R.J. Fallopian tube precursors of ovarian low- and high-grade serous neoplasms. Histopathology 2013, 62, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Flesken-Nikitin, A.; Hwang, C.I.; Cheng, C.Y.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [PubMed]
- Virant-Klun, I.; Zech, N.; Rozman, P.; Vogler, A.; Cvjeticanin, B.; Klemenc, P.; Malicev, E.; Meden-Vrtovec, H. Putative stem cells with an embryonic character isolated from the ovarian surface epithelium of women with no naturally present follicles and oocytes. Differentiation 2008, 76, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Bowen, N.J.; Walker, L.D.; Matyunina, L.V.; Logani, S.; Totten, K.A.; Benigno, B.B.; McDonald, J.F. Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med. Genom. 2009, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Jazedje, T.; Perin, P.M.; Czeresnia, C.E.; Maluf, M.; Halpern, S.; Secco, M.; Bueno, D.F.; Vieira, N.M.; Zucconi, E.; Zatz, M. Human fallopian tube: A new source of multipotent adult mesenchymal stem cells discarded in surgical procedures. J. Transl. Med. 2009, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levanon, K.; Ng, V.; Piao, H.Y.; Zhang, Y.; Chang, M.C.; Roh, M.H.; Kindelberger, D.W.; Hirsch, M.S.; Crum, C.P.; Marto, J.A.; et al. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene 2010, 29, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Barker, N. Ovary and fimbrial stem cells: Biology, niche and cancer origins. Nat. Rev. Mol. Cell Biol. 2015, 16, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. Pathogenesis of ovarian cancer: Lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Drapkin, R. Ovarian cancer pathogenesis: A model in evolution. J. Oncol. 2010, 2010, 932371. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.F.; Hu, W.; Sood, A.K. Microenvironment and pathogenesis of epithelial ovarian cancer. Horm. Cancer 2010, 1, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed]
- Birbeck, M.S.; Wheatley, D.N. An Electron Microscopic Study of the Invasion of Ascites Tumor Cells into the Abdominal Wall. Cancer Res. 1965, 25, 490–497. [Google Scholar] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.K. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol. Aspects Med. 2014, 39, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol. Oncol. 2009, 113, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Haviv, I.; Thompson, E.W. Soiling the seed: Microenvironment and epithelial mesenchymal plasticity. Cancer Microenviron. 2012, 5, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial—mesenchymal and mesenchymal—epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr. Cancer Drug Targets 2010, 10, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Maines-Bandiera, S.; Quinn, M.A.; Unger, W.G.; Dedhar, S.; Auersperg, N. Molecular pathways regulating EGF-induced epithelio-mesenchymal transition in human ovarian surface epithelium. Am. J. Physiol. Cell Physiol. 2006, 290, C1532–C1542. [Google Scholar] [CrossRef] [PubMed]
- Elloul, S.; Vaksman, O.; Stavnes, H.T.; Trope, C.G.; Davidson, B.; Reich, R. Mesenchymal-to-epithelial transition determinants as characteristics of ovarian carcinoma effusions. Clin. Exp. Metastasis 2010, 27, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.S. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: Functions and mechanisms. J. Biol. Chem. 2009, 284, 31493–31497. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.S.; Parks, W.C. Metalloproteinases: A parade of functions in matrix biology and an outlook for the future. Matrix Biol. 2015, 44–46, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tallant, C.; Marrero, A.; Gomis-Ruth, F.X. Matrix metalloproteinases: Fold and function of their catalytic domains. Biochim. Biophys. Acta 2010, 1803, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Sohail, A.; Sun, Q.; Zhao, H.; Bernardo, M.M.; Cho, J.A.; Fridman, R. MT4-(MMP17) and MT6-MMP (MMP25), A unique set of membrane-anchored matrix metalloproteinases: Properties and expression in cancer. Cancer Metastasis Rev. 2008, 27, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Naim, A.; Pan, Q.; Baig, M. Matrix Metalloproteinases (MMPs) in Liver Diseases. J. Clin. Exp. Hepatol. 2017, 7, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Gueders, M.M.; Foidart, J.-M.; Noel, A.; Cataldo, D.D. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: Potential implications in asthma and other lung diseases. Eur. J. Pharmacol. 2006, 533, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Groblewska, M.; Siewko, M.; Mroczko, B.; Szmitkowski, M. The role of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in the development of esophageal cancer. Folia Histochem. Cytobiol. 2012, 50, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M. Molecular determinants of metalloproteinase substrate specificity: Matrix metalloproteinase substrate binding domains, modules, and exosites. Mol. Biotechnol. 2002, 22, 51–86. [Google Scholar] [CrossRef]
- Xu, J.; Clark, R.A. A three-dimensional collagen lattice induces protein kinase C-zeta activity: Role in α2 integrin and collagenase mRNA expression. J. Cell Biol. 1997, 136, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wei, L.; Chen, Q.; Terek, R.M. CXCR4/SDF1 mediate hypoxia induced chondrosarcoma cell invasion through ERK signaling and increased MMP1 expression. Mol. Cancer 2010, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, S.; Lu, J.; Cao, Y.; Song, N.; Yang, T.; Dong, R.; Zang, L.; Yang, Y.; Wu, T.; et al. Correlation between MMP1-PAR1 axis and clinical outcome of primary gallbladder carcinoma. Jpn. J. Clin. Oncol. 2011, 41, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Sokolovic, A.; Sokolovic, M.; Boers, W.; Elferink, R.P.; Bosma, P.J. Insulin-like growth factor binding protein 5 enhances survival of LX2 human hepatic stellate cells. Fibrogenesis Tissue Repair 2010, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Orbe, J.; Paramo, J.A. Metalloproteases, vascular remodeling and atherothrombotic syndromes. Rev. Esp. Cardiol. 2007, 60, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Lemaître, V.; D’Armiento, J.; Okada, Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol. Int. 2010, 60, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Giebeler, N.; Zigrino, P. A Disintegrin and Metalloprotease (ADAM): Historical Overview of Their Functions. Toxins 2016, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.; Hundhausen, C.; Mentlein, R.; Schulte, A.; Berkhout, T.A.; Broadway, N.; Hartmann, D.; Sedlacek, R.; Dietrich, S.; Muetze, B.; et al. The transmembrane CXC-chemokine ligand 16 is induced by IFN-γ and TNF-α and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J. Immunol. 2004, 172, 6362–6372. [Google Scholar] [CrossRef] [PubMed]
- Dreymueller, D.; Pruessmeyer, J.; Groth, E.; Ludwig, A. The role of ADAM-mediated shedding in vascular biology. Eur. J. Cell Biol. 2012, 91, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, L.; Chi, C.; Wu, X.; Yang, X.; Han, M.; Xu, T.; Zhuang, Y.; Deng, K. ADAM10 is essential for early embryonic cardiovascular development. Dev. Dyn. 2010, 239, 2594–2602. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.M.; Sandhu, B.; Day, M.L. EGF promotes the shedding of soluble E-cadherin in an ADAM10-dependent manner in prostate epithelial cells. Cell Signal. 2012, 24, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Eichenauer, D.A.; Simhadri, V.L.; von Strandmann, E.P.; Ludwig, A.; Matthews, V.; Reiners, K.S.; von Tresckow, B.; Saftig, P.; Rose-John, S.; Engert, A.; et al. ADAM10 inhibition of human CD30 shedding increases specificity of targeted immunotherapy in vitro. Cancer Res. 2007, 67, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.H.; Ford, J.W.; Sturgill, J.L.; Gibb, D.R. CD23, an overlooked regulator of allergic disease. Curr. Allergy Asthma Rep. 2007, 7, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, P.C.; Boylan, K.L.M.; Walcheck, B.; Heinze, R.; Geller, M.A.; Argenta, P.A.; Skubitz, A.P.N. Ectodomain shedding of the cell adhesion molecule Nectin-4 in ovarian cancer is mediated by ADAM10 and ADAM17. J. Biol. Chem. 2017, 292, 6339–6351. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, P.J. Role of ADAM10 in intestinal crypt homeostasis and tumorigenesis. Biochim. Biophys. Acta 2017, 1864, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Tousseyn, T.; Thathiah, A.; Jorissen, E.; Raemaekers, T.; Konietzko, U.; Reiss, K.; Maes, E.; Snellinx, A.; Serneels, L.; Nyabi, O.; et al. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMs-9, ADAMs-15, and the γ-secretase. J. Biol. Chem. 2009, 284, 11738–11747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loechel, F.; Fox, J.W.; Murphy, G.; Albrechtsen, R.; Wewer, U.M. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem. Biophys. Res. Commun. 2000, 278, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.; Fischer, O.M.; Prenzel, N.; Zwick-Wallasch, E.; Schneider, M.; Hennighausen, L.; Ullrich, A. GPCR-induced migration of breast carcinoma cells depends on both EGFR signal transactivation and EGFR-independent pathways. Biol. Chem. 2005, 386, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Endsley, M.A.; Somasunderam, A.D.; Li, G.; Oezguen, N.; Thiviyanathan, V.; Murray, J.L.; Rubin, D.H.; Hodge, T.W.; O’Brien, W.A.; Lewis, B.; et al. Nuclear trafficking of the HIV-1 pre-integration complex depends on the ADAM10 intracellular domain. Virology 2014, 454–455, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Fourie, A.M.; Coles, F.; Moreno, V.; Karlsson, L. Catalytic activity of ADAM8, ADAM15, and MDC-L (ADAM28) on synthetic peptide substrates and in ectodomain cleavage of CD23. J. Biol. Chem. 2003, 278, 30469–30477. [Google Scholar] [CrossRef] [PubMed]
- Borrell-Pages, M.; Rojo, F.; Albanell, J.; Baselga, J.; Arribas, J. TACE is required for the activation of the EGFR by TGF-α in tumors. EMBO J. 2003, 22, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Canault, M.; Certel, K.; Schatzberg, D.; Wagner, D.D.; Hynes, R.O. The lack of ADAM17 activity during embryonic development causes hemorrhage and impairs vessel formation. PLoS ONE 2010, 5, e13433. [Google Scholar] [CrossRef] [PubMed]
- Allinson, T.M.; Parkin, E.T.; Turner, A.J.; Hooper, N.M. ADAMs family members as amyloid precursor protein α-secretases. J. Neurosci. Res. 2003, 74, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Arribas, J.; Esselens, C. ADAM17 as a therapeutic target in multiple diseases. Curr. Pharm. Des. 2009, 15, 2319–2335. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Avila, M.A. Amphiregulin. Semin. Cell Dev. Biol. 2014, 28, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, S.; Soejima, K.; Shimoda, M.; Abe, H.; Sasaki, A.; Okano, H.J.; Okano, H.; Okada, Y. Effect of ADAM28 on carcinoma cell metastasis by cleavage of von Willebrand factor. J. Natl. Cancer Inst. 2012, 104, 906–922. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E.; Song, H.; Hahm, C.; Yoon, S.Y.; Park, S.; Lee, H.R.; Hur, D.Y.; Kim, T.; Kim, C.H.; Bang, S.I.; et al. Expression of ADAM33 is a novel regulatory mechanism in IL-18-secreted process in gastric cancer. J. Immunol. 2009, 182, 3548–3555. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Charrier-Hisamuddin, L.; Laboisse, C.L.; Merlin, D. ADAM-15: A metalloprotease that mediates inflammation. FASEB J. 2008, 22, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, S.; Clark, I.M.; Kevorkian, L.; Edwards, D.R. The ADAMTS metalloproteinases. Biochem. J. 2005, 386, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; McKiernan, E.; O’Donovan, N.; McGowan, P.M. The role of ADAMs in disease pathophysiology. Clin. Chim. Acta 2009, 403, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Bischoff, R. Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: Biological function and structure. J. Proteome Res. 2011, 10, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Primakoff, P.; Myles, D.G. The ADAM gene family: Surface proteins with adhesion and protease activity. Trends Genet. 2000, 16, 83–87. [Google Scholar] [CrossRef]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Hougaard, S.; Loechel, F.; Xu, X.; Tajima, R.; Albrechtsen, R.; Wewer, U.M. Trafficking of human ADAM 12-L: Retention in the trans-Golgi network. Biochem. Biophys. Res. Commun. 2000, 275, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Howard, L.; Maciewicz, R.A.; Blobel, C.P. Cloning and characterization of ADAM28, evidence for autocatalytic pro-domain removal and for cell surface localization of mature ADAM28. Biochem. J. 2000, 348, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.F.; Courtneidge, S.A. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. ADAM proteins, their ligands, and clinical implications. Neurology 2012, 78, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAM10 regulates endothelial permeability and T cell transmigrstion by proteolysis of vascular endothelial cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Umata, T.; Hirata, M.; Takahashi, T.; Ryu, F.; Shida, S.; Takahashi, Y.; Tsuneoka, M.; Miura, Y.; Masuda, M.; Horiguchi, Y.; Mekada, E. A dual signaling cascade that regulates the ectodomain shedding of heparin-binding epidermal growth factor-like growth factor. J. Biol. Chem. 2001, 276, 30475–30482. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Shirakabe, K.; Werb, Z. The metalloprotease Kuzbanian (ADAM-10) mediates transactivation of EGF receptor by G protein-coupled receptors. J. Cell. Biol. 2002, 158, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Blobel, C.P. Ectodomain shedding of the EGF-receptor ligand eigen is mediated by ADAM-17. FEBS Lett. 2007, 581, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Hirata, M.; Yamazaki, A.; Kageyama, T.; Hasuwa, H.; Mizushima, H.; Tanaka, Y.; Yagi, H.; Sonoda, K.; Kai, M.; et al. Heparin-binding EGF-like growth factor is a promising target for ovarian cancer therapy. Cancer Res. 2004, 64, 8135–8145. [Google Scholar] [CrossRef] [PubMed]
- Rossello, A.; Nuti, E.; Ferrini, S.; Fabbi, M. Targeting ADAM17 Sheddase Activity in Cancer. Curr. Drug Targets. 2016, 17, 1908–1927. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, N.; Meyer, D.; Mauermann, A.; von der Heyde, J.; Wolf, J.; Schwarz, J.; Knittler, K.; Murphy, G.; Michalek, M.; Garbers, C.; et al. Shedding of Endogenous Interleukin-6 Receptor (IL-6R) Is Governed by A Disintegrin and Metalloproteinase (ADAM) Proteases while a Full-length IL-6R Isoform Localizes to Circulating Microvesicles. J. Biol. Chem. 2015, 290, 26059–26071. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Moreno, J.L.; Zandueta, C.; Montuenga, L.; Lecanda, F. Novel alternatively spliced ADAM8 isoforms contribute to the aggressive bone metastatic phenotype of lung cancer. Oncogene 2010, 29, 3758–3769. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Lu, D.; Scully, M.; Kakkar, V. ADAM proteins-therapeutic potential in cancer. Curr. Cancer Drug Targets 2008, 8, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Nath, D.; Slocombe, P.M.; Stephens, P.E.; Warn, A.; Hutchinson, G.R.; Yamada, K.M.; Docherty, A.J.; Murphy, G. Interaction of metargidin (ADAM-15) with αvβ3 and α5β1 integrins on different haemopoietic cells. J. Cell Sci. 1999, 112, 579–587. [Google Scholar] [PubMed]
- Byzova, T.V.; Goldman, C.K.; Pampori, N.; Thomas, K.A.; Bett, A.; Shattil, S.J.; Plow, E.F. A mechanism for modulation of cellular responses to VEGF: Activation of the integrins. Mol. Cell 2000, 6, 851–860. [Google Scholar] [CrossRef]
- McCabe, N.P.; De, S.; Vasanji, A.; Brainard, J.; Byzova, T.V. Prostate cancer specific integrin αvβ3 modulates bone metastatic growth and tissue remodeling. Oncogene 2007, 26, 6238–6243. [Google Scholar] [CrossRef] [PubMed]
- Wolfsberg, T.G.; Primakoff, P.; Myles, D.G.; White, J.M. ADAM, a novel family of membrane proteins containing a Disintegrin and Metalloprotease domain: Multipotential functions in cell-cell and cell-matrix interactions. J. Cell Biol. 1995, 131, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A.; Coppari, R.; De Franco, R.; Cho, J.Y.; Libermann, T.A.; Pinzani, M.; Toker, A. A secreted form of ADAM9 promotes carcinoma invasion through tumor-stromal interactions. Cancer Res. 2005, 65, 4728–4738. [Google Scholar] [CrossRef] [PubMed]
- Mechtersheimer, S.; Gutwein, P.; Agmon-Levin, N.; Stoeck, A.; Oleszewski, M.; Riedle, S.; Postina, R.; Fahrenholz, F.; Fogel, M.; Lemmon, V.; et al. Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J. Cell Biol. 2001, 155, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Karadag, A.; Zhou, M.; Croucher, P.I. ADAM-9 (MDC-9/meltrin-γ), a member of the a disintegrin and metalloproteinase family, regulates myeloma-cell-induced interleukin-6 production in osteoblasts by direct interaction with the αvβ5 integrin. Blood 2006, 107, 3271–3278. [Google Scholar] [CrossRef] [PubMed]
- Li, S.W.; Arita, M.; Fertala, A.; Bao, Y.; Kopen, G.C.; Langsjo, T.K.; Hyttinen, M.M.; Helminen, H.J.; Prockop, D.J. Transgenic mice with inactive alleles for procollagen N-proteinase (ADAMTS-2) develop fragile skin and male sterility. Biochem. J. 2001, 355, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, R.J.; Hirohata, S.; Engle, J.M.; Colige, A.; Cohn, D.H.; Eyre, D.R.; Apte, S.S. Procollagen II amino propeptide processing by ADAMTS-3. Insights on dermatosparaxis. J. Biol. Chem. 2001, 276, 31502–31509. [Google Scholar] [CrossRef] [PubMed]
- Bolz, H.; Ramirez, A.; von Brederlow, B.; Kubisch, C. Characterization of ADAMTS14, a novel member of the ADAMTS metalloproteinase family. Biochim. Biophys. Acta 2001, 1522, 221–225. [Google Scholar] [CrossRef]
- Colige, A.; Vandenberghe, I.; Thiry, M.; Lambert, C.A.; Van Beeumen, J.; Li, S.W.; Prockop, D.J.; Lapiere, C.M.; Nusgens, B.V. Cloning and characterization of ADAMTS-14, a novel ADAMTS displaying high homology with ADAMTS-2 and ADAMTS-3. J. Biol. Chem. 2002, 277, 5756–5766. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Manzaneque, J.C.; Westling, J.; Thai, S.N.; Luque, A.; Knauper, V.; Murphy, G.; Sandy, J.D.; Iruela-Arispe, M.L. ADAMTS1 cleaves aggrecan at multiple sites and is differentially inhibited by metalloproteinase inhibitors. Biochem. Biophys. Res. Commun. 2002, 293, 501–508. [Google Scholar] [CrossRef]
- Abbaszade, I.; Liu, R.Q.; Yang, F.; Rosenfeld, S.A.; Ross, O.H.; Link, J.R.; Ellis, D.M.; Tortorella, M.D.; Pratta, M.A.; Hollis, J.M.; et al. Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J. Biol. Chem. 1999, 274, 23443–23450. [Google Scholar] [CrossRef] [PubMed]
- Somerville, R.P.; Longpre, J.M.; Jungers, K.A.; Engle, J.M.; Ross, M.; Evanko, S.; Wight, T.N.; Leduc, R.; Apte, S.S. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J. Biol. Chem. 2003, 278, 9503–9513. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, M.D.; Burn, T.C.; Pratta, M.A.; Abbaszade, I.; Hollis, J.M.; Liu, R.; Rosenfeld, S.A.; Copeland, R.A.; Decicco, C.P.; Wynn, R.; et al. Purification and cloning of aggrecanase-1, a member of the ADAMTS family of proteins. Science 1999, 284, 1664–1666. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, C.; Xu, X.; Zhu, X.; Dai, D. Downregulation of A disintegrin and metallopeptidase with thrombospondin motif type 1 by DNA hypermethylation in human gastric cancer. Mol. Med. Rep. 2015, 12, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Colige, A.; Nuytinck, L.; Hausser, I.; van Essen, A.J.; Thiry, M.; Herens, C.; Ades, L.C.; Malfait, F.; Paepe, A.D.; Franck, P.; et al. Novel types of mutation responsible for the dermatosparactic type of Ehlers-Danlos syndrome (Type VIIC) and common polymorphisms in the ADAMTS2 gene. J. Invest. Dermatol. 2004, 123, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Dubail, J.; Apte, S.S. Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics. Matrix Biol. 2015, 44–46, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Bekhouche, M.; Colige, A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015, 44–46, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, M.; Bonetti, S.; Boselli, M.L.; Miccoli, R.; Trabetti, E.; Malerba, G.; Pignatti, P.F.; Bonora, E.; Del Prato, S.; Bonadonna, R.C. PPARG2 Pro12Ala and ADAMTS9 rs4607103 as “insulin resistance loci” and “insulin secretion loci” in Italian individuals. The GENFIEV study and the Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 4. Acta Diabetol. 2013, 50, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Akyol, O.; Akyol, S.; Chen, C.H. Update on ADAMTS13 and VWF in cardiovascular and hematological disorders. Clin. Chim. Acta 2016, 463, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Ogata, A.; Kang, S.; Ebina, K.; Shi, K.; Nojima, S.; Kimura, T.; Ito, D.; Morimoto, K.; Nishide, M.; et al. Semaphorin 4D Contributes to Rheumatoid Arthritis by Inducing Inflammatory Cytokine Production: Pathogenic and Therapeutic Implications. Arthritis Rheumatol. 2015, 67, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Tsuzaka, K.; Itami, Y.; Takeuchi, T.; Shinozaki, N.; Morishita, T. ADAMTS5 is a biomarker for prediction of response to infliximab in patients with rheumatoid arthritis. J. Rheumatol. 2010, 37, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Du, C.; Wang, H.; Zhang, C. Increased serum ADAMTS-4 in knee osteoarthritis: A potential indicator for the diagnosis of osteoarthritis in early stages. Genet. Mol. Res. 2014, 13, 9642–9649. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, D.; Yiotakis, A. Specific targeting of metzincin family members with small-molecule inhibitors: Progress toward a multifarious challenge. Bioorg. Med. Chem. 2008, 16, 8781–8794. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sayans García, M.; Suarez-Penaranda, J.M.; Gayoso-Diz, P.; Barros-Angueira, F.; Gandara-Rey, J.M.; Garcia-Garcia, A. Tissue inhibitor of metalloproteinases in oral squamous cell carcinomas—A therapeutic target? Cancer Lett. 2012, 323, 11–19. [Google Scholar]
- Abbenante, G.; Fairlie, D.P. Protease inhibitors in the clinic. Med. Chem. 2005, 1, 71–104. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.T. Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail. Rev. 2004, 9, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Lopez-Otin, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A. Tumor necrosis factor-α converting enzyme. Int. J. Biochem. Cell Biol. 2002, 34, 1–5. [Google Scholar] [CrossRef]
- Mezyk, R.; Bzowska, M.; Bereta, J. Structure and functions of tumor necrosis factor-α converting enzyme. Acta Biochim. Pol. 2003, 50, 625–645. [Google Scholar] [PubMed]
- Grootveld, M.; McDermott, M. BMS-561392. Bristol-Myers Squibb. Curr. Opin. Investig. Drugs 2003, 4, 598–602. [Google Scholar] [PubMed]
- Turner, S.L.; Blair-Zajdel, M.E.; Bunning, R.A. ADAMs and ADAMTSs in cancer. Br. J. Biomed. Sci. 2009, 66, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ling, Y.; Zhang, C.; Xu, Y.; Gao, L.; Li, R.; Zhu, J.; Fan, L.; Wei, L. The silencing of RECK gene is associated with promoter hypermethylation and poor survival in hepatocellular carcinoma. Int. J. Biol. Sci. 2012, 8, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.J.; Wu, D.C.; Cheng, K.H.; Chen, L.T.; Hung, W.C. RECK inhibits stemness gene expression and tumorigenicity of gastric cancer cells by suppressing ADAM-mediated Notch1 activation. J. Cell. Physiol. 2014, 229, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.A.; Roy, D.M.; Reyngold, M.; Giri, D.; Snyder, A.; Turcan, S.; Badwe, C.R.; Lyman, J.; Bromberg, J.; King, T.A.; et al. RECK controls breast cancer metastasis by modulating a convergent, STAT3-dependent neoangiogenic switch. Oncogene 2015, 34, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, J.; Xu, Z.Y.; Xie, H.L. Expression of RECK and matrix metalloproteinase-2 in ameloblastoma. BMC Cancer 2009, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, H.F.; Zhang, H.Z. Expression of RECK and MMPs in Hepatoblastoma and Neuroblastoma and Comparative Analysis on the Tumor Metastasis. Asian Pac. J. Cancer Prev. 2015, 16, 4007–4011. [Google Scholar] [CrossRef] [PubMed]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef] [PubMed]
- DeClerck, Y.A. Tissue Inhibitors of Metalloproteinases in cancer. In Cancer Metastasis—Biology and Treatment; Foidart, J.M., Muschel, R.J., Eds.; Springer: Dordrecht, Netherlands, 2002; Volume 4. [Google Scholar]
- Radisky, E.S.; Raeeszadeh-Sarmazdeh, M.; Radisky, D.C. Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. J. Cell. Biochem. 2017, 118, 3531–3548. [Google Scholar] [CrossRef] [PubMed]
- Langton, K.P.; Barker, M.D.; McKie, N. Localization of the functional domains of human tissue inhibitor of metalloproteinases-3 and the effects of a Sorsby’s fundus dystrophy mutation. J. Biol. Chem. 1998, 273, 16778–16781. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.C.; Langley, K.E.; Mendiaz, E.A.; Parker, V.P.; Taylor, S.M.; DeClerck, Y.A. The C-terminal domain of tissue inhibitor of metalloproteinases-2 is required for cell binding but not for antimetalloproteinase activity. Biochem. Biophys. Res. Commun. 1997, 236, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Chirco, R.; Liu, X.W.; Jung, K.K.; Kim, H.R. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 2006, 25, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Koskivirta, I.; Kassiri, Z.; Rahkonen, O.; Kiviranta, R.; Oudit, G.Y.; McKee, T.D.; Kytö, V.; Saraste, A.; Jokinen, E.; Liu, P.P.; Vuorio, E.; Khokha, R. Mice with tissue inhibitor of metalloproteinases 4 (Timp4) deletion succumb to induced myocardial infarction but not to cardiac pressure overload. J. Biol. Chem. 2010, 285, 24487–24493. [Google Scholar] [CrossRef] [PubMed]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Adamson, A.; Ghoreschi, K.; Rittler, M.; Chen, Q.; Sun, H.W.; Vahedi, G.; Kanno, Y.; Stetler-Stevenson, W.G.; O’Shea, J.J.; Laurence, A. Tissue inhibitor of metalloproteinase 1 is preferentially expressed in Th1 and Th17 T-helper cell subsets and is a direct STAT target gene. PLoS ONE 2013, 8, e59367. [Google Scholar] [CrossRef] [PubMed]
- Hyc, A.; Osiecka-Iwan, A.; Niderla-Bielinska, J.; Moskalewski, S. Influence of LPS, TNF, TGF-ss1 and IL-4 on the expression of MMPs, TIMPs and selected cytokines in rat synovial membranes incubated in vitro. Int. J. Mol. Med. 2011, 27, 127–137. [Google Scholar] [PubMed]
- Wang, K.; Lin, B.; Brems, J.J.; Gamelli, R.L. Hepatic apoptosis can modulate liver fibrosis through TIMP1 pathway. Apoptosis 2013, 18, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. The tumor microenvironment: Regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2. Cancer Metastasis Rev. 2008, 27, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Remillard, T.C.; Bratslavsky, G.; Jensen-Taubman, S.; Stetler-Stevenson, W.G.; Bourboulia, D. Molecular mechanisms of tissue inhibitor of metalloproteinase 2 in the tumor microenvironment. Mol. Cell. Ther. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Fussbroich, B.; Wagener, N.; Macher-Goeppinger, S.; Benner, A.; Falth, M.; Sultmann, H.; Holzer, A.; Hoppe-Seyler, K.; Hoppe-Seyler, F. EZH2 depletion blocks the proliferation of colon cancer cells. PLoS ONE 2011, 6, e21651. [Google Scholar] [CrossRef] [PubMed]
- Bourboulia, D.; Han, H.; Jensen-Taubman, S.; Gavil, N.; Isaac, B.; Wei, B.; Neckers, L.; Stetler-Stevenson, W.G. TIMP-2 modulates cancer cell transcriptional profile and enhances E-cadherin/β-catenin complex expression in A549 lung cancer cells. Oncotarget 2013, 4, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guo, Y.; Dong, R.; Dai, R.; Zhou, M. Suppressor of cytokine signaling 1-modulated metalloproteinases and tissue inhibitor of metalloproteinase in pulmonary fibrosis. Mol. Med. Rep. 2015, 12, 3855–3861. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.H.; Ebrahem, Q.; Ali, M.; Cutler, A.; Bell, B.; Prayson, N.; Sears, J.; Knauper, V.; Murphy, G.; Anand-Apte, B. Tissue inhibitor of metalloproteinases-3 peptides inhibit angiogenesis and choroidal neovascularization in mice. PLoS ONE 2013, 8, e55667. [Google Scholar] [CrossRef] [PubMed]
- El Mabrouk, M.; Qureshi, H.Y.; Li, W.Q.; Sylvester, J.; Zafarullah, M. Interleukin-4 antagonizes oncostatin M and transforming growth factor β-induced responses in articular chondrocytes. J. Cell. Biochem. 2008, 103, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, K.; Han, X.; Mao, C.; Zhang, K.; Zhao, T.; Zhao, J. The imbalance between TIMP3 and matrix-degrading enzymes plays an important role in intervertebral disc degeneration. Biochem. Biophys. Res. Commun. 2016, 469, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Adissu, H.A.; McKerlie, C.; Di Grappa, M.; Waterhouse, P.; Xu, Q.; Fang, H.; Khokha, R.; Wood, G.A. Timp3 loss accelerates tumour invasion and increases prostate inflammation in a mouse model of prostate cancer. Prostate 2015, 75, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Fogarasi, M.; Janssen, A.; Weber, B.H.; Stohr, H. Molecular dissection of TIMP3 mutation S156C associated with Sorsby fundus dystrophy. Matrix Biol. 2008, 27, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. Tissue inhibitors of metalloproteinases in cell signaling: Metalloproteinase-independent biological activities. Sci. Signal. 2008, 1, re6. [Google Scholar] [CrossRef] [PubMed]
- Taube, M.E.; Liu, X.W.; Fridman, R.; Kim, H.R. TIMP-1 regulation of cell cycle in human breast epithelial cells via stabilization of p27KIP1 protein. Oncogene 2006, 25, 3041–3048. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, M.L.; Stetler-Stevenson, W.G. Tissue inhibitor of metalloproteinase-2 stimulates fibroblast proliferation via a cAMP-dependent mechanism. J. Biol. Chem. 1995, 270, 13453–13459. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Suzuki, M.; Iwata, H.; Koike, T.; Hamaguchi, M.; Shinagawa, A.; Noguchi, T.; Hayakawa, T. Tyrosine phosphorylation is crucial for growth signaling by tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2). FEBS Lett. 1996, 396, 103–107. [Google Scholar] [CrossRef]
- Wang, T.; Yamashita, K.; Iwata, K.; Hayakawa, T. Both tissue inhibitors of metalloproteinases-1 (TIMP-1) and TIMP-2 activate Ras but through different pathways. Biochem. Biophys. Res. Commun. 2002, 296, 201–205. [Google Scholar] [CrossRef]
- Stetler-Stevenson, W.G.; Gavil, N.V. Normalization of the tumor microenvironment: Evidence for tissue inhibitor of metalloproteinase-2 as a cancer therapeutic. Connect. Tissue Res. 2014, 55, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.W.; Li, H.; Guedez, L.; Wingfield, P.T.; Diaz, T.; Salloum, R.; Wei, B.Y.; Stetler-Stevenson, W.G. TIMP-2 mediated inhibition of angiogenesis: An MMP-independent mechanism. Cell 2003, 114, 171–180. [Google Scholar] [CrossRef]
- Seo, D.W.; Li, H.; Qu, C.K.; Oh, J.; Kim, Y.S.; Diaz, T.; Wei, B.; Han, J.W.; Stetler-Stevenson, W.G. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J. Biol. Chem. 2006, 281, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.A.; Roy, R.; Lee, S.; Yang, J.; Panigrahy, D.; Van Vliet, K.J.; Moses, M.A. The anti-angiogenic peptide, loop 6, binds insulin-like growth factor-1 receptor. J. Biol. Chem. 2010, 285, 41886–41895. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.K.; Liu, X.W.; Chirco, R.; Fridman, R.; Kim, H.R. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006, 25, 3934–3942. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, L.; Jaworski, D.M. Tissue inhibitor of metalloproteinase-2 promotes neuronal differentiation by acting as an anti-mitogenic signal. J. Neurosci. 2005, 25, 4917–4929. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Kita, D.; Futami, K.; Yamashita, J.; Fujimoto, N.; Sato, H.; Okada, Y. Roles of membrane type 1 matrix metalloproteinase and tissue inhibitor of metalloproteinases 2 in invasion and dissemination of human malignant glioma. J. Neurosurg. 2001, 94, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Kachra, Z.; Beaulieu, E.; Delbecchi, L.; Mousseau, N.; Berthelet, F.; Moumdjian, R.; Del Maestro, R.; Beliveau, R. Expression of matrix metalloproteinases and their inhibitors in human brain tumors. Clin. Exp. Metastasis 1999, 17, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yang, Z.; Zhang, H.; Gan, M.; Zhou, T.; Wang, S. The expression and clinical significance of matrix metalloproteinase 7 and tissue inhibitor of matrix metalloproteinases 2 in clear cell renal cell carcinoma. Exp. Ther. Med. 2013, 5, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Suemitsu, R.; Yoshino, I.; Tomiyasu, M.; Fukuyama, S.; Okamoto, T.; Maehara, Y. Serum tissue inhibitors of metalloproteinase-1 and -2 in patients with non-small cell lung cancer. Surg. Today 2004, 34, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Honkavuori-Toivola, M.; Talvensaari-Mattila, A.; Soini, Y.; Turpeenniemi-Hujanen, T.; Santala, M. Immunoreactivity for TIMP-2 is associated with a favorable prognosis in endometrial carcinoma. Tumour Biol. 2012, 33, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.A.; Cepeda, M.A.; Damjanovski, S. Analysis of the MMP-dependent and independent functions of tissue inhibitor of metalloproteinase-2 on the invasiveness of breast cancer cells. J. Cell Commun. Signal. 2012, 6, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, H.S.; Kim, T.H.; Lee, J.S.; Lee, S.T.; Lee, S.J. Growth-stimulatory activity of TIMP-2 is mediated through c-Src activation followed by activation of FAK, PI3-kinase/AKT, and ERK1/2 independent of MMP inhibition in lung adenocarcinoma cells. Oncotarget 2015, 6, 42905–42922. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Dou, C.; Jia, Y.; Tu, K.; Zheng, X. TIMP-1 activated carcinoma-associated fibroblasts inhibit tumor apoptosis by activating SDF1/CXCR4 signaling in hepatocellular carcinoma. Oncotarget 2015, 6, 12061–12079. [Google Scholar] [CrossRef] [PubMed]
- Alpizar-Alpizar, W.; Laerum, O.D.; Christensen, I.J.; Ovrebo, K.; Skarstein, A.; Hoyer-Hansen, G.; Ploug, M.; Illemann, M. Tissue Inhibitor of Metalloproteinase-1 Is Confined to Tumor-Associated Myofibroblasts and Is Increased With Progression in Gastric Adenocarcinoma. J. Histochem. Cytochem. 2016, 64, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Jiang, Y.; Chen, Y.R.; Zheng, H.C.; Zeng, W.; Li, Y.Y.; Yin, A.; Nie, Y. Expression and inhibitory role of TIMP-3 in hepatocellular carcinoma. Oncol. Rep. 2016, 36, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Wu, Y.; Stack, M.S. Ovarian cancer-associated proteinases. Cancer Treat. Res. 2002, 107, 331–351. [Google Scholar] [PubMed]
- Zhang, Y.; Chen, Q. Relationship between matrix metalloproteinases and the occurrence and development of ovarian cancer. Braz. J. Med. Biol. Res. 2017, 50, e6104. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Xu, S.; Xu, Y.; Ma, J.; Li, J.; Xu, P. The expression of tumor-derived and stromal-derived matrix metalloproteinase 2 predicted prognosis of ovarian cancer. Int. J. Gynecol. Cancer 2015, 25, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Perigny, M.; Bairati, I.; Harvey, I.; Beauchemin, M.; Harel, F.; Plante, M.; Tetu, B. Role of immunohistochemical overexpression of matrix metalloproteinases MMP-2 and MMP-11 in the prognosis of death by ovarian cancer. Am. J. Clin. Pathol. 2008, 129, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Ishikura, H.; Kawarada, Y.; Ogawa, Y.; Sakuragi, N.; Fujimoto, S.; Yoshiki, T. Expression of matrix metalloproteinases and related tissue inhibitors in the cyst fluids of ovarian mucinous neoplasms. Gynecol. Oncol. 2000, 78, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jin, X.; Lin, D.; Liu, Z.; Zhang, X.; Lu, Y.; Liu, Y.; Wang, M.; Yang, M.; Li, J.; et al. Clinicopathologic significance of claudin-6, occludin, and matrix metalloproteinases-2 expression in ovarian carcinoma. Diagn. Pathol. 2013, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Desmeules, P.; Trudel, D.; Turcotte, S.; Sirois, J.; Plante, M.; Gregoire, J.; Renaud, M.C.; Orain, M.; Tetu, B.; Bairati, I. Prognostic significance of TIMP-2, MMP-2, and MMP-9 on high-grade serous ovarian carcinoma using digital image analysis. Hum. Pathol. 2015, 46, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Brun, J.L.; Cortez, A.; Lesieur, B.; Uzan, S.; Rouzier, R.; Darai, E. Expression of MMP-2, -7, -9, MT1-MMP and TIMP-1 and -2 has no prognostic relevance in patients with advanced epithelial ovarian cancer. Oncol. Rep. 2012, 27, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Halon, A.; Nowak-Markwitz, E.; Donizy, P.; Matkowski, R.; Maciejczyk, A.; Gansukh, T.; Gyorffy, B.; Spaczynski, M.; Zabel, M.; Lage, H.; et al. Enhanced immunoreactivity of TIMP-2 in the stromal compartment of tumor as a marker of favorable prognosis in ovarian cancer patients. J. Histochem. Cytochem. 2012, 60, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B. Ovarian carcinoma and serous effusions. Changing views regarding tumor progression and review of current literature. Anal. Cell. Pathol. 2001, 23, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Brun, J.L.; Cortez, A.; Commo, F.; Uzan, S.; Rouzier, R.; Darai, E. Serous and mucinous ovarian tumors express different profiles of MMP-2, -7, -9, MT1-MMP, and TIMP-1 and -2. Int. J. Oncol. 2008, 33, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, D.R.; Simoens, C.; Bogers, J.P.; Murta, E.F.; Michelin, M.A. Angiogenesis Markers in Gynecological Tumors and Patents for Anti-Angiogenic Approach: Review. Recent Pat. Anticancer Drug Discov. 2015, 10, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.H.; Wang, S.T.; Yao, M.Z.; Cai, J.H.; Chen, C.Y.; Yang, Z.X.; Hong, L.; Yang, S.Y. Screening of the residual normal ovarian tissue adjacent to orthotopic epithelial ovarian carcinomas in nude mice. Genet. Mol. Res. 2014, 13, 2978–2986. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Goldberg, I.; Gotlieb, W.H.; Kopolovic, J.; Ben-Baruch, G.; Nesland, J.M.; Berner, A.; Bryne, M.; Reich, R. High levels of MMP-2, MMP-9, MT1-MMP and TIMP-2 mRNA correlate with poor survival in ovarian carcinoma. Clin. Exp. Metastasis 1999, 17, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kasberg, W.C.; Celo, A.; Liang, Z.; Quispe, K.; Stack, M.S. Post-translational modification of the membrane type 1 matrix metalloproteinase (MT1-MMP) cytoplasmic tail impacts ovarian cancer multicellular aggregate dynamics. J. Biol. Chem. 2017, 292, 13111–13121. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Reich, R.; Berner, A.; Givant-Horwitz, V.; Goldberg, I.; Risberg, B.; Kristensen, G.B.; Trope, C.G.; Bryne, M.; Kopolovic, J.; et al. Ovarian carcinoma cells in serous effusions show altered MMP-2 and TIMP-2 mRNA levels. Eur. J. Cancer 2001, 37, 2040–2049. [Google Scholar] [CrossRef]
- Winiarski, B.K.; Cope, N.; Alexander, M.; Pilling, L.C.; Warren, S.; Acheson, N.; Gutowski, N.J.; Whatmore, J.L. Clinical Relevance of Increased Endothelial and Mesothelial Expression of Proangiogenic Proteases and VEGFA in the Omentum of Patients with Metastatic Ovarian High-Grade Serous Carcinoma. Transl. Oncol. 2014, 7, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xiao, Y.J.; Singh, L.S.; Zhao, X.; Zhao, Z.; Feng, L.; Rose, T.M.; Prestwich, G.D.; Xu, Y. Lysophosphatidic acid is constitutively produced by human peritoneal mesothelial cells and enhances adhesion, migration, and invasion of ovarian cancer cells. Cancer Res. 2006, 66, 3006–3014. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Fishman, D.A.; Liu, Y.; Ellerbroek, S.M.; Stack, M.S. Lysophosphatidic acid promotes matrix metalloproteinase (MMP) activation and MMP-dependent invasion in ovarian cancer cells. Cancer Res. 2001, 61, 3194–3199. [Google Scholar] [PubMed]
- Burkhalter, R.J.; Westfall, S.D.; Liu, Y.; Stack, M.S. Lysophosphatidic Acid Initiates Epithelial to Mesenchymal Transition and Induces β-Catenin-mediated Transcription in Epithelial Ovarian Carcinoma. J. Biol. Chem. 2015, 290, 22143–22154. [Google Scholar] [CrossRef] [PubMed]
- Ray, U.; Roy, S.S.; Chowdhury, S.R. Lysophosphatidic Acid Promotes Epithelial to Mesenchymal Transition in Ovarian Cancer Cells by Repressing SIRT1. Cell Physiol. Biochem. 2017, 41, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Gil, O.D.; Lee, C.; Ariztia, E.V.; Wang, F.Q.; Smith, P.J.; Hope, J.M.; Fishman, D.A. Lysophosphatidic acid (LPA) promotes E-cadherin ectodomain shedding and OVCA429 cell invasion in an uPA-dependent manner. Gynecol. Oncol. 2008, 108, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burkhalter, R.; Symowicz, J.; Chaffin, K.; Ellerbroek, S.; Stack, M.S. Lysophosphatidic Acid disrupts junctional integrity and epithelial cohesion in ovarian cancer cells. J. Oncol. 2012, 2012, 501492. [Google Scholar] [CrossRef] [PubMed]
- Cowden Dahl, K.D.; Symowicz, J.; Ning, Y.; Gutierrez, E.; Fishman, D.A.; Adley, B.P.; Stack, M.S.; Hudson, L.G. Matrix metalloproteinase 9 is a mediator of epidermal growth factor-dependent e-cadherin loss in ovarian carcinoma cells. Cancer Res. 2008, 68, 4606–4613. [Google Scholar] [CrossRef] [PubMed]
- Symowicz, J.; Adley, B.P.; Gleason, K.J.; Johnson, J.J.; Ghosh, S.; Fishman, D.A.; Hudson, L.G.; Stack, M.S. Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res. 2007, 67, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Veatch, A.L.; Carson, L.F.; Ramakrishnan, S. Differential expression of the cell-cell adhesion molecule E-cadherin in ascites and solid human ovarian tumor cells. Int. J. Cancer 1994, 58, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Sundfeldt, K.; Ivarsson, K.; Rask, K.; Haeger, M.; Hedin, L.; Brannstrom, M. Higher levels of soluble E-cadherin in cyst fluid from malignant ovarian tumours than in benign cysts. Anticancer Res. 2001, 21, 65–70. [Google Scholar] [PubMed]
- Mahner, S.; Woelber, L.; Eulenburg, C.; Schwarz, J.; Carney, W.; Jaenicke, F.; Milde-Langosch, K.; Mueller, V. TIMP-1 and VEGF-165 serum concentration during first-line therapy of ovarian cancer patients. BMC Cancer 2010, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Bachvarov, D.; L’Esperance, S.; Popa, I.; Bachvarova, M.; Plante, M.; Tetu, B. Gene expression patterns of chemoresistant and chemosensitive serous epithelial ovarian tumors with possible predictive value in response to initial chemotherapy. Int. J. Oncol. 2006, 29, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Ryner, L.; Guan, Y.; Firestein, R.; Xiao, Y.; Choi, Y.; Rabe, C.; Lu, S.; Fuentes, E.; Huw, L.Y.; Lackner, M.R.; et al. Upregulation of Periostin and Reactive Stroma Is Associated with Primary Chemoresistance and Predicts Clinical Outcomes in Epithelial Ovarian Cancer. Clin. Cancer Res. 2015, 21, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Tressel, S.L.; Kaimal, R.; Balla, M.; Lam, F.H.; Covic, L.; Kuliopulos, A. Identification of a metalloprotease-chemokine signaling system in the ovarian cancer microenvironment: Implications for antiangiogenic therapy. Cancer Res. 2010, 70, 5880–5890. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Q.; Fisher, J.; Fishman, D.A. MMP-1-PAR1 axis mediates LPA-induced epithelial ovarian cancer (EOC) invasion. Gynecol. Oncol. 2011, 120, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Abi Saab W, F.; Modi, N.; Stewart, A.; Liu, J.; Chadee, D.N. Mixed lineage kinase 3 is required for matrix metalloproteinase expression and invasion in ovarian cancer cells. Exp. Cell Res. 2012, 318, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Q.; So, J.; Reierstad, S.; Fishman, D.A. Matrilysin (MMP-7) promotes invasion of ovarian cancer cells by activation of progelatinase. Int. J. Cancer 2005, 114, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Chen, C.A.; Chen, P.J.; Chiang, Y.C.; Chen, Y.L.; Mao, T.L.; Lin, H.W.; Lin Chiang, W.H.; Cheng, W.F. Mesothelin enhances invasion of ovarian cancer by inducing MMP-7 through MAPK/ERK and JNK pathways. Biochem. J. 2012, 442, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Ellerbroek, S.M.; Hudson, L.G.; Stack, M.S. Proteinase requirements of epidermal growth factor-induced ovarian cancer cell invasion. Int. J. Cancer 1998, 78, 331–337. [Google Scholar] [CrossRef]
- Choi, J.H.; Choi, K.C.; Auersperg, N.; Leung, P.C. Gonadotropins activate proteolysis and increase invasion through protein kinase A and phosphatidylinositol 3-kinase pathways in human epithelial ovarian cancer cells. Cancer Res. 2006, 66, 3912–3920. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Wu, Y.N.; Wang, S.L.; Lin, Q.H.; He, M.F.; Liu, Q.L.; Wang, J.H. Docosahexaenoic Acid Modulates Invasion and Metastasis of Human Ovarian Cancer via Multiple Molecular Pathways. Int. J. Gynecol. Cancer 2016, 26, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Yang, Y.; Cui, L.; Yang, J.; Li, X.; Yang, Y.; Duan, H. Bisdemethoxycurcumin inhibits ovarian cancer via reducing oxidative stress mediated MMPs expressions. Sci. Rep. 2016, 6, 28773. [Google Scholar] [CrossRef] [PubMed]
- Dolo, V.; D’Ascenzo, S.; Giusti, I.; Millimaggi, D.; Taraboletti, G.; Pavan, A. Shedding of membrane vesicles by tumor and endothelial cells. Ital. J. Anat. Embryol. 2005, 110 (Suppl. 1), 127–133. [Google Scholar] [PubMed]
- Tanaka, Y.; Miyamoto, S.; Suzuki, S.O.; Oki, E.; Yagi, H.; Sonoda, K.; Yamazaki, A.; Mizushima, H.; Maehara, Y.; Mekada, E.; et al. Clinical significance of heparin-binding epidermal growth factor-like growth factor and a disintegrin and metalloprotease 17 expression in human ovarian cancer. Clin. Cancer Res. 2005, 11, 4783–4792. [Google Scholar] [CrossRef] [PubMed]
- Vlad, C.; Kubelac, P.; Onisim, A.; Irimie, A.; Achimas-Cadariu, P. The role of CDCP1 (CUB domain-containing protein 1) and ADAM12 (a disintegrin and metalloproteinase 12) in ovarian cancer. J. BUON 2015, 20, 673–679. [Google Scholar] [PubMed]
- Yasukawa, M.; Liu, Y.; Hu, L.; Cogdell, D.; Gharpure, K.M.; Pradeep, S.; Nagaraja, A.S.; Sood, A.K.; Zhang, W. ADAMTS16 mutations sensitize ovarian cancer cells to platinum-based chemotherapy. Oncotarget 2016, 8, 88410–88420. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Greening, D.; Samardzija, C.; Escalona, R.M.; Chen, M.; Findlay, J.K.; Kannourakis, G. Unique proteome signature of post-chemotherapy ovarian cancer ascites-derived tumor cells. Sci. Rep. 2016, 6, 30061. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype of chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.A.; Dos Santos, L.; Turri, J.A.; Nonogaki, S.; Buim, M.; Lima, J.F.; de Jesus Viana Pinheiro, J.; Bueno de Toledo Osorio, C.A.; Soares, F.A.; Freitas, V.M. Prognostic Value of ADAMTS Proteases and Their Substrates in Epithelial Ovarian Cancer. Pathobiology 2016, 83, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A collagen-remodeling gene signature regulated by TGF-β signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Schiemann, W.P. The TGF-β paradox in human cancer: An update. Future Oncol. 2009, 5, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–177. [Google Scholar] [PubMed]
- Sritananuwat, P.; Sueangoen, N.; Thummarati, P.; Islam, K.; Suthiphongchai, T. Blocking ERK1/2 signaling impairs TGF-β1 tumor promoting function but enhances its tumor suppressing role in intrahepatic cholangiocarcinoma cells. Cancer Cell Int. 2017, 17, 85. [Google Scholar] [CrossRef] [PubMed]
- Alsina-Sanchís, E.; Figueras, A.; Lahiguera, A.; Gil-Martín, M.; Pardo, B.; Piulats, J.M.; Martí, L.; Ponce, J.; Matias-Guiu, X.; Vidal, A.; Villanueva, A.; Viñals, F. TGFβ controls ovarian cancer cell proliferation. Int. J. Mol. Sci. 2017, 18, 1658. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell. Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, K.; Luwor, R.B.; Zhu, H.; McNally, O.; Quinn, M.A.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 2014, 14, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latifi, A.; Escalona, R.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Distinct molecular signature of recurrent ovarian tumor cells isolated from the ascites of advanced-stage serous ovarian cancer patients. J. Cancer Stem Cell Res. 2015, 2, e1006. [Google Scholar] [CrossRef]
- Chan, E.; Luwor, R.; Burns, C.; Kannourakis, G.; Findlay, J.; Ahmed, N. Momelotinib decreased tumor burden and prolonged disease-free remission period in a mouse model of ovarian cancer. 2018; submitted. [Google Scholar]
- Valacca, C.; Tassone, E.; Mignatti, P. TIMP-2 Interaction with MT1-MMP Activates the AKT Pathway and Protects Tumor Cells from Apoptosis. PLoS ONE 2015, 10, e0136797. [Google Scholar] [CrossRef] [PubMed]
- Samardzija, C.; Luwor, R.B.; Volchek, M.; Quinn, M.A.; Findlay, J.K.; Ahmed, N. A critical role of Oct4A in mediating metastasis and disease-free survival in a mouse model of ovarian cancer. Mol. Cancer 2015, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Kveiborg, M.; Jacobsen, J.; Lee, M.H.; Nagase, H.; Wewer, U.M.; Murphy, G. Selective inhibition of ADAM12 catalytic activity through engineering of tissue inhibitor of metalloproteinase 2 (TIMP-2). Biochem. J. 2010, 430, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Atkinson, S.; Rapti, M.; Handsley, M.; Curry, V.; Edwards, D.; Murphy, G. The activity of a designer tissue inhibitor of metalloproteinases (TIMP)-1 against native membrane type 1 matrix metalloproteinase (MT1-MMP) in a cell-based environment. Cancer Lett. 2010, 290, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.X.; Rapti, M.; Tsigkou, A.; Lee, M.H. Expanding the Activity of Tissue Inhibitors of Metalloproteinase (TIMP)-1 against Surface-Anchored Metalloproteinases by the Replacement of Its C-Terminal Domain: Implications for Anti-Cancer Effects. PLoS ONE 2015, 10, e0136384. [Google Scholar] [CrossRef] [PubMed]
- Vadikolia, C.M.; Tsatalas, C.; Anagnostopoulos, K.; Trypsianis, G.; Pantelidou, D.; Bazdiara, I.; Anastasiadis, A.; Spanoudakis, E.; Kotsianidis, I.; Margaritis, D.; et al. Proteolytic matrix metallopeptidases and inhibitors in BCR-ABL1-negative myeloproliferative neoplasms: Correlation with JAK2 mutation status. Acta Haematol. 2011, 126, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhong, B.; Chen, M.; Yang, L.; Yang, G.; Li, Y.; Wang, H.; Wang, G.; Li, W.; Cui, J.; et al. Epigenetic reprogramming reverses the malignant epigenotype of the MMP/TIMP axis genes in tumor cells. Int. J. Cancer 2014, 134, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Miyake, H.; Nishikawa, M.; Tei, H.; Furukawa, J.; Harada, K.; Fujisawa, M. Significance of circulating matrix metalloproteinase-9 to tissue inhibitor of metalloproteinases-2 ratio as a predictor of disease progression in patients with metastatic renal cell carcinoma receiving sunitinib. Urol. Oncol. 2014, 32, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Hoegy, S.E.; Oh, H.R.; Corcoran, M.L.; Stetler-Stevenson, W.G. Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. J. Biol. Chem. 2001, 276, 3203–3214. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Cho, Y.R.; Kim, H.J.; Oh, J.S.; Ahn, E.K.; Ko, H.J.; Hwang, B.J.; Lee, S.J.; Cho, Y.; Kim, Y.K.; et al. Antagonism of VEGF-A-induced increase in vascular permeability by an integrin α3β1-Shp-1-cAMP/PKA pathway. Blood 2012, 120, 4892–4902. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Tsang, P.S.; Diaz, T.M.; Wei, B.Y.; Stetler-Stevenson, W.G. TIMP-2 modulates VEGFR-2 phosphorylation and enhances phosphodiesterase activity in endothelial cells. Lab. Investig. 2010, 90, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Bourboulia, D.; Jensen-Taubman, S.; Rittler, M.R.; Han, H.Y.; Chatterjee, T.; Wei, B.; Stetler-Stevenson, W.G. Endogenous angiogenesis inhibitor blocks tumor growth via direct and indirect effects on tumor microenvironment. Am. J. Pathol. 2011, 179, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Batra, J.; Robinson, J.; Mehner, C.; Hockla, A.; Miller, E.; Radisky, D.C.; Radisky, E.S. PEGylation extends circulation half-life while preserving in vitro and in vivo activity of tissue inhibitor of metalloproteinases-1 (TIMP-1). PLoS ONE 2012, 7, e50028. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Chanda, D.; Cody, J.J.; Rivera, A.A.; Waehler, R.; Siegal, G.P.; Douglas, J.T.; Ponnazhagan, S. Conditionally replicating adenovirus expressing TIMP2 increases survival in a mouse model of disseminated ovarian cancer. PLoS ONE 2011, 6, e25131. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Cody, J.J.; Rivera, A.A.; Waehler, R.; Wang, M.; Kimball, K.J.; Alvarez, R.A.; Siegal, G.P.; Douglas, J.T.; Ponnazhagan, S. Conditionally replicating adenovirus expressing TIMP2 for ovarian cancer therapy. Clin. Cancer Res. 2011, 17, 538–549. [Google Scholar] [CrossRef] [PubMed]


 ) control SKOV3, OVCAR5 and HEY cell lines after treatment with IC50 doses of (
) control SKOV3, OVCAR5 and HEY cell lines after treatment with IC50 doses of (  ) paclitaxel (0.67 µg/mL, 0.002 µg/mL and 0.0004 µg/mL respectively) and (
) paclitaxel (0.67 µg/mL, 0.002 µg/mL and 0.0004 µg/mL respectively) and (  ) cisplatin (3.95 µg/mL, 4.57 µg/mL and 1.19 µg/mL respectively) for 72 h by qRT-PCR as described previously [235,238]. SKOV-3, OVCAR-5 and HEY cell lines have been described before [238]. The relative expression of gene of interest was normalized to house-keeping 18S gene. Data are shown as the mean ± SEM (n = 3). ** p < 0.01, *** p < 0.001. (B) The expression of ERCC1 and TUBB3A was performed in control, paclitaxel and cisplatin-treated ovarian cancer cell lines as described in Figure 3A. ** p < 0.01, **** p < 0.0001.
) control SKOV3, OVCAR5 and HEY cell lines after treatment with IC50 doses of ( ) paclitaxel (0.67 µg/mL, 0.002 µg/mL and 0.0004 µg/mL respectively) and ( ) cisplatin (3.95 µg/mL, 4.57 µg/mL and 1.19 µg/mL respectively) for 72 h by qRT-PCR as described previously [235,238]. SKOV-3, OVCAR-5 and HEY cell lines have been described before [238]. The relative expression of gene of interest was normalized to house-keeping 18S gene. Data are shown as the mean ± SEM (n = 3). ** p < 0.01, *** p < 0.001. (B) The expression of ERCC1 and TUBB3A was performed in control, paclitaxel and cisplatin-treated ovarian cancer cell lines as described in Figure 3A. ** p < 0.01, **** p < 0.0001.
) cisplatin (3.95 µg/mL, 4.57 µg/mL and 1.19 µg/mL respectively) for 72 h by qRT-PCR as described previously [235,238]. SKOV-3, OVCAR-5 and HEY cell lines have been described before [238]. The relative expression of gene of interest was normalized to house-keeping 18S gene. Data are shown as the mean ± SEM (n = 3). ** p < 0.01, *** p < 0.001. (B) The expression of ERCC1 and TUBB3A was performed in control, paclitaxel and cisplatin-treated ovarian cancer cell lines as described in Figure 3A. ** p < 0.01, **** p < 0.0001.
) control SKOV3, OVCAR5 and HEY cell lines after treatment with IC50 doses of ( ) paclitaxel (0.67 µg/mL, 0.002 µg/mL and 0.0004 µg/mL respectively) and ( ) cisplatin (3.95 µg/mL, 4.57 µg/mL and 1.19 µg/mL respectively) for 72 h by qRT-PCR as described previously [235,238]. SKOV-3, OVCAR-5 and HEY cell lines have been described before [238]. The relative expression of gene of interest was normalized to house-keeping 18S gene. Data are shown as the mean ± SEM (n = 3). ** p < 0.01, *** p < 0.001. (B) The expression of ERCC1 and TUBB3A was performed in control, paclitaxel and cisplatin-treated ovarian cancer cell lines as described in Figure 3A. ** p < 0.01, **** p < 0.0001.

 chemoresistant cells
chemoresistant cells  cancer cells.
chemoresistant cells cancer cells.
cancer cells.
chemoresistant cells cancer cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Gene | Inhibited by | Location | Targets | References |
|---|---|---|---|---|---|
| Collagenases | MMP-1 | TIMP-1 TIMP-2 TIMP-3 TIMP-4 | secreted | Substrates include Col I, II, III, VII, VIII and X; gelatin, MMP-2, MMP-9, proteoglycans, aggrecan, casein, serpins, versican, proTNF-α, IGF-BP-3, IGF-BP-5, IL-1β, SDF-1, PAR1, CD49b, binds α2β1-integrin | [43,44,45,46,47,48,49,50,51,52,53] |
| MMP-8 | TIMP-1 TIMP-2 TIMP-4 | secreted | Substrates include Col I, II, III, VII, VIII, X, aggrecan, gelatin, laminin, nidogen, proteoglycans, ADAMTS-1, pro-MMP-8, proTNF-α, α2-antiplasmin, IGF-BP. | [43,44,45,46,47] | |
| MMP-13 | TIMP-1 TIMP-2 TIMP-3 TIMP-4 | secreted | Substrates include Col I, II, III, IV, IX, X, XIV, gelatin, laminin, proteoglycans, aggrecans, fibronectin, pro TNF-α, pro-MMP-9 and pro-MMP-13, SDF-1. | [43,44,45,46,47,48,49,50,51,52,53] | |
| Gelatinases | MMP-2 | TIMP-1 TIMP-2 TIMP-3 TIMP-4 | secreted | Substrates include Gelatin, Col I, II, III, IV, V, VII, X, XI, XIV, fibronectin, laminin-5, elastin, aggrecan, versican, active MMP-9, active MMP-13, SDF-1, IGF-BPs, IL-1β, pro-TGF-β1,CXCL6, α1-antyproteinase. | [43,44,45,46,47,53] |
| MMP-9 | TIMP-1 TIMP-2 TIMP-3 TIMP-4 | secreted | Substrates include Gelatin, Col IV, V, VII, X, elastin, laminin, versican, aggrecan, fibronectin, vitronectin, CXCL5, IL-1β, pro-TGF-β1, plasminogen, pro-TNF-α, SDF-1, CXCL6. | [43,44,45,46,47,53] | |
| Stromelysins | MMP-3 | TIMP-1 TIMP-2 TIMP-3 TIMP-4 | Secreted | Substrates include Col II, III, IV, IX, X, XI, gelatin, fibronectin, laminin, proteoglycan, versican, pro-MMP-1, pro-MMP-7, pro-MMP-8, pro-MMP-9, pro-MMP-13, pro-TNF-α, E-cadherin, l-selectin, SDF-1, pro-HB-EGF, pro-IL-1β. | [45,46,47] |
| MMP-10 | TIMP-1 TIMP-2 | Secreted | Substrates include Col I, III, IV, V, laminin, casein, pro-MMP-1, pro-MMP-8, pro-MMP-10, fibronectin, proteoglycans, elastin, gelatin | [45,46,47] | |
| MMP-11 | TIMP-1 | secreted | Substrates include Col IV, fibronectin, laminin, gelatin, aggrecan, α1-antitrypsin, α1-proteinase inhibitor, IGF-BP-1 | [45,46,47,51] | |
| Matrylisin | MMP-7 | TIMP-1 TIMP-2 TIMP-3 TIMP-4 | Secreted | Substrates include: Col I, IV, X, fibronectin, laminin, gelatin, elastin, aggrecan, casein, proteoglycans, pro-MMP-1, pro-MMP-2, pro-MMP-7, pro-MMP-8, pro-TNF-α, E-cadherin, Fas-L, β4 integrin | [45,46,47,48,49,50,51,52,53] |
| MMP-26 | TIMP-4 | secreted | substrates include: Col I, IV, fibrogen, fibronectin, laminin, gelatin, casein, elastin, proteoglycans, pro-MMP-2, pro-TNF-α, E-cadherin | [45,54] | |
| Membrane Type-MMPs | MMP-14 | TIMP-2 TIMP-3 TIMP-4 | membrane-associated | Substrates include: Col I, II, III, gelatin, fibronectin, laminin, vitronectin, proteoglycans, tenascin, pro-MMP-2, pro-MMP-13, SDF-1, pro-TNF-α, αvβ3 integrin | [43,44,45,46,47,48,49,50,51,52,53] |
| MMP-15 | TIMP-2 TIMP-3 TIMP-4 | membrane-associated | Substrates include gelatin, Col I, fibronectin, laminin, nidogen, tenascin, pro-MMP-2, pro-MMP-13, pro-TNF-α | [43,44,45,46,47,48,49,50,51,52,53] | |
| MMP-16 | TIMP-2 TIMP-4 | membrane-associated | substrates include Col I, gelatin, fibronectin, laminin, vitronectin, aggrecan, casein, pro-MMP-2, pro-MMP-13 | [43,44,45,46,47,48,49,50,51,52,53] | |
| Metalloelastase | MMP-12 | TIMP-1 TIMP-4 | secreted | Substrates include elastin, fibronectin, Col IV, gelatin, proteoglycans, plasminogen, laminin | [43,44,45,46,47,48,49,50,51,52,53] |
| Other MMPs | MMP-19 | TIMP-2 TIMP-4 | – | Substrates include: Col I, IV, gelatin, aggrecan, casein, tenascin, nidogen, laminin. | [47] |
| Active ADAMs | ADAM-10 | TIMP-1 TIMP-3 | membrane-associated | integrins CD11b/CD18 (Mac-1), junctional adhesion molecule (JAM)-A; Notch;
EGFR ligands: HB-EGF; EGF and betacellulin; APP; Notch2, Notch3, N-, and E-cadherin; CD23, CD30, CD44, DLL1, Fas-L, HER2, L1, TNF-α Notch receptor, ephrin A5, collagen IV., CXCL16, nectin-4 | [55,56,57,58,59,60,61,62,63,64] |
| ADAM-12 | TIMP-1 (weak) TIMP-2 TIMP-3 | membrane-associated | gelatinase; HB-EGF; collagen IV, DLL1, fibronectin; IGF-BP3, IGF-BP5, ADAM-10, Betacellulin | [55,58,65,66,67] | |
| ADAM-15 | TIMP-3 | membrane-associated | EGFR ligands: HB-EGF; amphiregulin, CD23, collagen IV, E-cadherin, ADAM-10, gelatin | [55,60,68,69,70] | |
| ADAM-17 | TIMP-3 TIMP-4 | membrane-associated | TACE (TNF-α converting enzyme); Mac-1, JAM-A; EGFR ligands, HB-EGF and amphiregulin; APP; EpCAM and ErbB4; CD44, collagen XVII, DLL1, epiregulin, epigen, ICAM-1, L-selectin, Notch1, transforming growth factor α (TGF-α), TNF-α, V-CAM1; mucin-1 (MUC-1), tumor necrosis factor receptor (TNFR)I, TNFRII, IL-6R, HER-4, vascular endothelial growth factor receptor 2 (VEGFR2), nectin-4 | [55,63,71,72,73,74,75,76] | |
| ADAM-28 | TIMP-3 TIMP-4 | membrane-associated | IGBP3, VWF, CD23 | [55,76] | |
| ADAM-33 | TIMP-3 TIMP-4 | membrane-associated | IL-18, APP, KL-1, insulin B chain | [55,77,78,79] | |
| ADAMTS | ADAMTS1 | TIMP-2 TIMP-3 | secreted | Substrates include: Aggrecan, versican, syndecan 4, TFPI-2, semaphorin 3C, nidogen-1, -2, desmocollin-3, dystroglycan, mac-2, gelatin (denatured collagen type I), amphiregulin, TGF-α, heparin-binding EGF | [80,81] |
| ADAMTS-2 | TIMP-3 (weak) | secreted | Substrates include: Fibrillar procollagens types I-III and V | [80,81] | |
| ADAMTS-4 | TIMP-1 TIMP-3 TIMP-4 | secreted | Substrates include: Aggrecan, versican, reelin, biglycan, brevican, matrilin-3, α2-macroglobulin, Cartilage oligomeric protein (COMP) | [80,81] | |
| ADAMTS-5 | TIMP-3 TIMP-4 (weak) | secreted | Substrates include: Aggrecan, versican, reelin, biglycan, matrilin-4, brevican, α2-macroglobulin | [80,81] | |
| ADAMTS-9 | TIMP-3 | secreted | Substrates include: Aggrecan, versican | [80,81] |
| Characteristic | TIMP-1 | TIMP-2 | TIMP-3 | TIMP-4 |
|---|---|---|---|---|
| Mol. weight (kDa) | 28 | 21 | 24 | 22 |
| Chromosome (human) | X11p11.23–11.4 | 17q23–25 | 22q12.1–q13.2 | 3q25 |
| mRNA (kb) | 0.9 | 1.2, 1.7, 3.5 | 2.4, 2.8, 4.5 | 1.4 |
| Glycosylation | Yes | No | Partial | No |
| Amino acids (mature protein) | 184 | 194 | 188 | 194 |
| Binding | Pro-MMP-9, cell surface | Pro-MMP-2, cell surface | ECM, Pro-MMP-9 and pro-MMP2 | Pro-MMP-2 |
| Apoptotic effects | Negative (Suppresses) | Positive(Stimulates) Negative (Protects) | Positive (Enhances) | Positive(Stimulates) Negative (Protects) |
| Cell Growth | Stimulates | Stimulates Suppresses | Suppresses | No effect |
| Angiogenesis | Mediate through interaction with β1 integrin and CD63 | Negative | Negative | Negative |
| Other Genes known to block | VEGFR2, VEGF | |||
| Down-regulated by | STAT3, IL-4 | EZH2, SOCS1, Baicalein (drug), Rosuvastatin (cholesterol lowering drug) | TNF, IL-4, | TGF-β1, LPS, TNF (weak), IL-4 (weak) |
| Upregulated by | TGF-β1, LPS, c-Jun | TGF-β1, TET1, angiotensin-II | TGF-β1, LPS | |
| References | [144,145,146,147,148,149] | [144,146,148,150,151,152,153,154] | [144,145,146,148,154,155,156,157,158,159] | [144,145,146,147,148] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escalona, R.M.; Chan, E.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. The Many Facets of Metzincins and Their Endogenous Inhibitors: Perspectives on Ovarian Cancer Progression. Int. J. Mol. Sci. 2018, 19, 450. https://doi.org/10.3390/ijms19020450
Escalona RM, Chan E, Kannourakis G, Findlay JK, Ahmed N. The Many Facets of Metzincins and Their Endogenous Inhibitors: Perspectives on Ovarian Cancer Progression. International Journal of Molecular Sciences. 2018; 19(2):450. https://doi.org/10.3390/ijms19020450
Chicago/Turabian StyleEscalona, Ruth M., Emily Chan, George Kannourakis, Jock K. Findlay, and Nuzhat Ahmed. 2018. "The Many Facets of Metzincins and Their Endogenous Inhibitors: Perspectives on Ovarian Cancer Progression" International Journal of Molecular Sciences 19, no. 2: 450. https://doi.org/10.3390/ijms19020450
APA StyleEscalona, R. M., Chan, E., Kannourakis, G., Findlay, J. K., & Ahmed, N. (2018). The Many Facets of Metzincins and Their Endogenous Inhibitors: Perspectives on Ovarian Cancer Progression. International Journal of Molecular Sciences, 19(2), 450. https://doi.org/10.3390/ijms19020450