The Impact of Aging on Cardio and Cerebrovascular Diseases

 ,
,  ,
, 
 , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
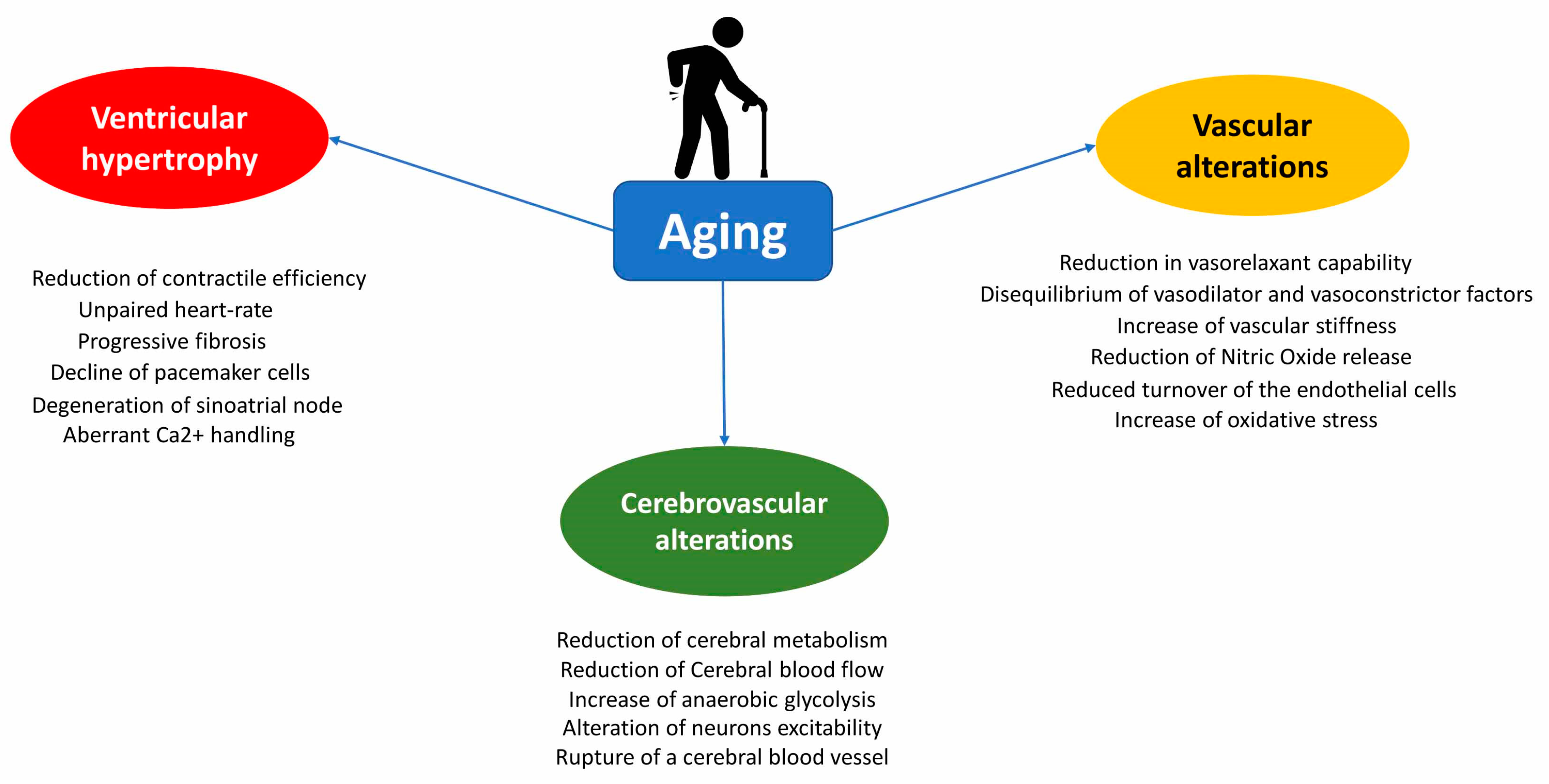
2. Structural and Physiological Changes in Heart during Aging
3. Vascular Alterations during Aging
4. Decline of Endothelial Vasorelaxation during Aging
5. Sources of Oxidative Stress and Vascular Aging
6. Age-Related Cerebrovascular System Modifications
7. Brain Stroke
8. Genetic Impact on Aging
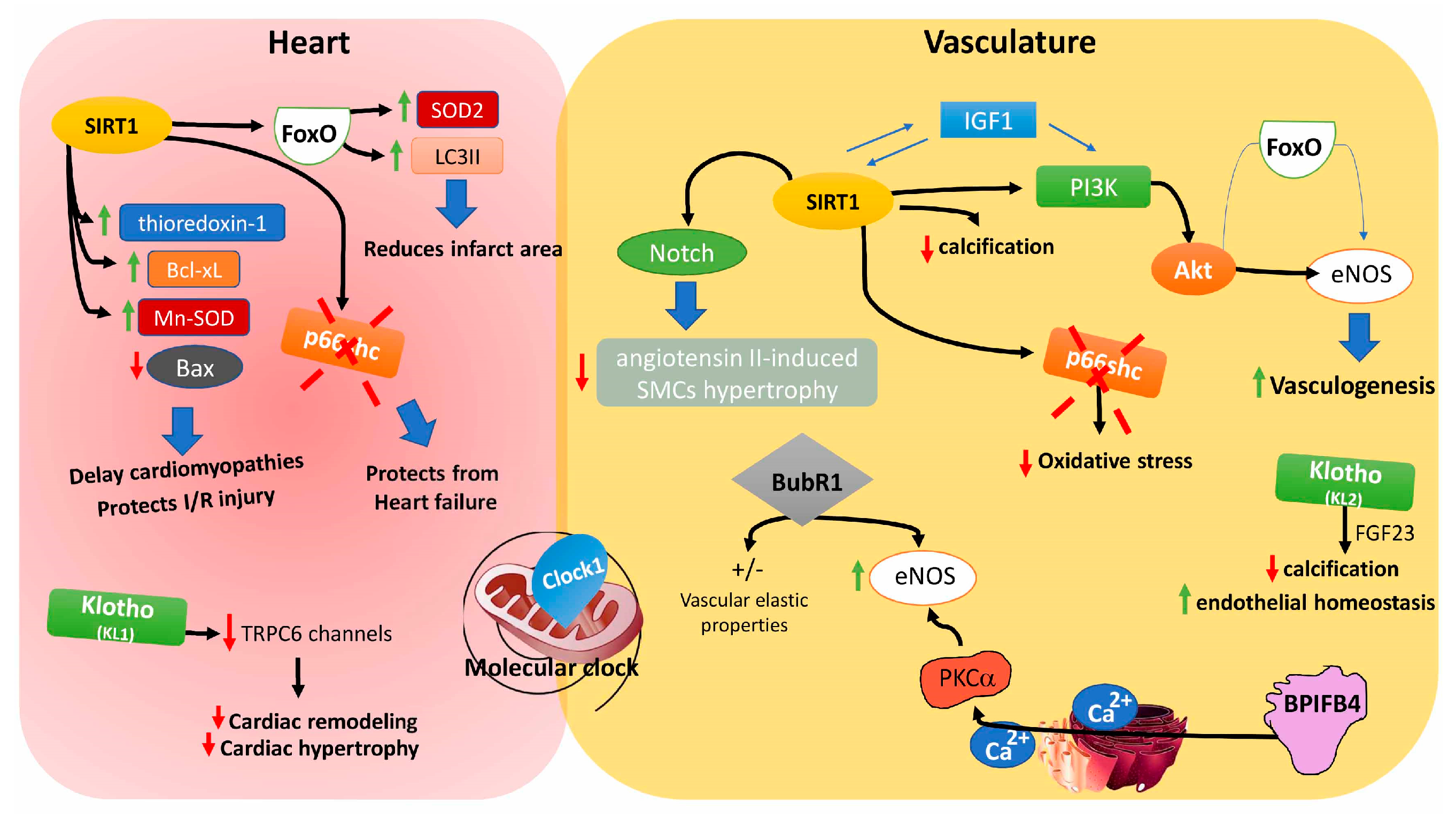
8.1. Sirtuins
8.2. Insulin-Like Growth Factors
8.3. Forkhead Box Proteins
8.4. Clock 1
8.5. p66shc
8.6. Klotho Gene
8.7. BubR1
8.8. Bactericidal/Permeability-Increasing Family B Member 4
8.9. The Role of Telomeres in the Aging
9. Conclusions
Conflicts of Interest
References
- Gerstenblith, G.; Frederiksen, J.; Yin, F.C.; Fortuin, N.J.; Lakatta, E.G.; Weisfeldt, M.L. Echocardiographic assessment of a normal adult aging population. Circulation 1977, 56, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Melissari, M.; Capasso, J.M.; Anversa, P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ. Res. 1991, 68, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Hees, P.S.; Fleg, J.L.; Lakatta, E.G.; Shapiro, E.P. Left ventricular remodeling with age in normal men versus women: Novel insights using three-dimensional magnetic resonance imaging. Am. J. Cardiol. 2002, 90, 1231–1236. [Google Scholar] [CrossRef]
- Goor, D.; Lillehei, C.W.; Edwards, J.E. The “sigmoid septum”. Variation in the contour of the left ventricular outt. Am. J. Roentgenol. 1969, 107, 366–376. [Google Scholar] [CrossRef]
- Rodeheffer, R.J.; Gerstenblith, G.; Becker, L.C.; Fleg, J.L.; Weisfeldt, M.L.; Lakatta, E.G. Exercise cardiac output is maintained with advancing age in healthy human subjects: Cardiac dilatation and increased stroke volume compensate for a diminished heart rate. Circulation 1984, 69, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Kawamato, A.; Matsubayashi, K.; Nishinaga, M.; Kimura, S.; Kuzume, O.; Chikamori, T.; Yamada, M.; Takada, J.; Kitazumi, T.; et al. Normal and abnormal aging of cardiovascular system. Nihon Ronen Igakkai Zasshi 1991, 28, 302–307. [Google Scholar] [PubMed]
- Schulman, S.P.; Lakatta, E.G.; Fleg, J.L.; Lakatta, L.; Becker, L.C.; Gerstenblith, G. Age-related decline in left ventricular filling at rest and exercise. Am. J. Physiol. 1992, 263, H1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.R.; Grossman, S.J.; Schectman, K.B.; Biello, D.R.; Ludbrook, P.A.; Ehsani, A.A. Left ventricular diastolic filling and its association with age. Am. J. Cardiol. 1986, 58, 531–535. [Google Scholar] [CrossRef]
- Fleg, J.L.; O’Connor, F.; Gerstenblith, G.; Becker, L.C.; Clulow, J.; Schulman, S.P.; Lakatta, E.G. Impact of age on the cardiovascular response to dynamic upright exercise in healthy men and women. J. Appl. Physiol. 1995, 78, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Spina, R.J.; Turner, M.J.; Ehsani, A.A. β-adrenergic-mediated improvement in left ventricular function by exercise training in older men. Am. J. Physiol. 1998, 274, H397–H404. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.T.; Wenzelburger, F.; Lee, E.; Heatlie, G.; Leyva, F.; Patel, K.; Frenneaux, M.; Sanderson, J.E. The pathophysiology of heart failure with normal ejection fraction: Exercise echocardiography reveals complex abnormalities of both systolic and diastolic ventricular function involving torsion, untwist, and longitudinal motion. J. Am. Coll. Cardiol. 2009, 54, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, K.V.; Sanna, S.; Scuteri, A.; Strait, J.B.; Orru, M.; Parsa, A.; Lin, P.I.; Maschio, A.; Lai, S.; Piras, M.G.; et al. Col4a1 is associated with arterial stiffness by genome-wide association scan. Circ. Cardiovasc. Genet. 2009, 2, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Julius, S.; Amery, A.; Whitlock, L.S.; Conway, J. Influence of age on the hemodynamic response to exercise. Circulation 1967, 36, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Gerstenblith, G.; Angell, C.S.; Shock, N.W.; Weisfeldt, M.L. Prolonged contraction duration in aged myocardium. J. Clin. Investig. 1975, 55, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Yin, F.C. Myocardial aging: Functional alterations and related cellular mechanisms. Am. J. Physiol. 1982, 242, H927–H941. [Google Scholar] [CrossRef] [PubMed]
- Pugh, K.G.; Wei, J.Y. Clinical implications of physiological changes in the aging heart. Drugs Aging 2001, 18, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Craft, N.; Schwartz, J.B. Effects of age on intrinsic heart rate, heart rate variability, and AV conduction in healthy humans. Am. J. Physiol. 1995, 268, H1441–H1452. [Google Scholar] [CrossRef] [PubMed]
- Fleg, J.L.; Das, D.N.; Wright, J.; Lakatta, E.G. Age-associated changes in the components of atrioventricular conduction in apparently healthy volunteers. J. Gerontol. 1990, 45, M95–100. [Google Scholar] [CrossRef] [PubMed]
- Golden, G.S.; Golden, L.H. The “nona” electrocardiogram: Findings in 100 patients of the 90 plus age group. J. Am. Geriatr. Soc. 1974, 22, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Harlan, W.R., Jr.; Graybiel, A.; Mitchell, R.E.; Oberman, A.; Osborne, R.K. Serial electrocardiograms: Their reliability and prognostic validity during a 24-yr. Period. J. Chronic Dis. 1967, 20, 853–867. [Google Scholar] [CrossRef]
- Mihalick, M.J.; Fisch, C. Electrocardiographic findings in the aged. Am. Heart J. 1974, 87, 117–128. [Google Scholar] [CrossRef]
- Manolio, T.A.; Furberg, C.D.; Rautaharju, P.M.; Siscovick, D.; Newman, A.B.; Borhani, N.O.; Gardin, J.M.; Tabatznik, B. Cardiac arrhythmias on 24-h ambulatory electrocardiography in older women and men: The cardiovascular health study. J. Am. Coll. Cardiol. 1994, 23, 916–925. [Google Scholar] [CrossRef]
- Fleg, J.L.; Kennedy, H.L. Cardiac arrhythmias in a healthy elderly population: Detection by 24-hour ambulatory electrocardiography. Chest 1982, 81, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Fisch, C. Introduction: Chronic ventricular arrhythmias—A major unresolved health problem. Heart Lung 1981, 10, 451–454. [Google Scholar] [PubMed]
- Lakatta, E.G.; Sollott, S.J. Perspectives on mammalian cardiovascular aging: Humans to molecules. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2002, 132, 699–721. [Google Scholar] [CrossRef]
- Lakatta, E.G. Age-associated cardiovascular changes in health: Impact on cardiovascular disease in older persons. Heart Fail. Rev. 2002, 7, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Sollott, S.J.; Pepe, S. The old heart: Operating on the edge. Novartis Found. Symp. 2001, 235, 172–196, discussion 196–201, 217–120. [Google Scholar] [PubMed]
- Lakatta, E.G. Cardiovascular regulatory mechanisms in advanced age. Physiol. Rev. 1993, 73, 413–467. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Deficient neuroendocrine regulation of the cardiovascular system with advancing age in healthy humans. Circulation 1993, 87, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Esler, M.D.; Turner, A.G.; Kaye, D.M.; Thompson, J.M.; Kingwell, B.A.; Morris, M.; Lambert, G.W.; Jennings, G.L.; Cox, H.S.; Seals, D.R. Aging effects on human sympathetic neuronal function. Am. J. Physiol. 1995, 268, R278–R285. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part ii: The aging heart in health: Links to heart disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Cigola, E.; Kajstura, J.; Li, B.; Meggs, L.G.; Anversa, P. Angiotensin ii activates programmed myocyte cell death in vitro. Exp. Cell Res. 1997, 231, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Sadoshima, J.; Xu, Y.; Slayter, H.S.; Izumo, S. Autocrine release of angiotensin ii mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 1993, 75, 977–984. [Google Scholar] [CrossRef]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Kaley, G.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Mechanisms of vascular aging: New perspectives. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Zieman, S.J.; Melenovsky, V.; Kass, D.A. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Avolio, A.P.; Mergner, W.J.; Robinowitz, M.; Herderick, E.E.; Cornhill, J.F.; Guo, S.Y.; Liu, T.H.; Ou, D.Y.; O’Rourke, M. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am. J. Pathol. 1991, 139, 1119–1129. [Google Scholar] [PubMed]
- Farasat, S.M.; Morrell, C.H.; Scuteri, A.; Ting, C.T.; Yin, F.C.; Spurgeon, H.A.; Chen, C.H.; Lakatta, E.G.; Najjar, S.S. Pulse pressure is inversely related to aortic root diameter implications for the pathogenesis of systolic hypertension. Hypertension 2008, 51, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part i: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y. Aging and arterial-cardiac interactions in the elderly. Int. J. Cardiol. 2012, 155, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, S.E. Ageing of the conduit arteries. J. Pathol. 2007, 211, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Tschudi, M.R.; Barton, M.; Bersinger, N.A.; Moreau, P.; Cosentino, F.; Noll, G.; Malinski, T.; Luscher, T.F. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J. Clin. Investig. 1996, 98, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Cernadas, M.R.; Sanchez de Miguel, L.; Garcia-Duran, M.; Gonzalez-Fernandez, F.; Millas, I.; Monton, M.; Rodrigo, J.; Rico, L.; Fernandez, P.; de Frutos, T.; et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 1998, 83, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Wang, M.; Lakatta, E.G.; Ungvari, Z. Inflammation and endothelial dysfunction during aging: Role of NF-κB. J. Appl. Physiol. 2008, 105, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, A.; Tesauro, M.; Rizza, S.; Iantorno, M.; Federici, M.; Lauro, D.; Campia, U.; Turriziani, M.; Fusco, A.; Cocciolillo, G.; et al. Endothelial function and arterial stiffness in normotensive normoglycemic first-degree relatives of diabetic patients are independent of the metabolic syndrome. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Manas, L.; El-Assar, M.; Vallejo, S.; Lopez-Doriga, P.; Solis, J.; Petidier, R.; Montes, M.; Nevado, J.; Castro, M.; Gomez-Guerrero, C.; et al. Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 2009, 8, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Briet, M.; Boutouyrie, P. Large and small artery cross-talk and recent morbidity-mortality trials in hypertension. Hypertension 2009, 54, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Edelberg, J.M.; Reed, M.J. Aging and angiogenesis. Front. Biosci. 2003, 8, s1199–s1209. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K.; Stringer, S.E.; Alexander, M.Y. Endothelial progenitor cells enter the aging arena. Front. Physiol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Wang, Y.; Yang, Z.; Tu, C.; Xu, M.G.; Wang, J.M. Circulating endothelial progenitor cell deficiency contributes to impaired arterial elasticity in persons of advancing age. J. Hum. Hypertens. 2006, 20, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Hoeber, S.; Froese, S.; Klink, I.; Stichtenoth, D.O.; Galuppo, P.; Jakob, M.; Tsikas, D.; Anker, S.D.; Poole-Wilson, P.A.; et al. Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circ. Res. 2007, 100, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.; Keymel, S.; Niesler, U.; Ziemann, J.; Kelm, M.; Kalka, C. Impaired progenitor cell activity in age-related endothelial dysfunction. J. Am. Coll. Cardiol. 2005, 45, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Roach, M.R.; Burton, A.C. The effect of age on the elasticity of human iliac arteries. Can. J. Biochem. Physiol. 1959, 37, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden-Spek, J.J.; Staessen, J.A.; Fagard, R.H.; Hoeks, A.P.; Boudier, H.A.; van Bortel, L.M. Effect of age on brachial artery wall properties differs from the aorta and is gender dependent: A population study. Hypertension 2000, 35, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Mattei, P.; Ghiadoni, L.; Gennari, A.; Fasolo, C.B.; Sudano, I.; Salvetti, A. Aging and endothelial function in normotensive subjects and patients with essential hypertension. Circulation 1995, 91, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Egashira, K.; Inou, T.; Hirooka, Y.; Kai, H.; Sugimachi, M.; Suzuki, S.; Kuga, T.; Urabe, Y.; Takeshita, A. Effects of age on endothelium-dependent vasodilation of resistance coronary artery by acetylcholine in humans. Circulation 1993, 88, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.L.; Sumner, D.J.; McLean, K.; Reid, J.L. Effect of age on the responsiveness of vascular α-adrenoceptors in man. J. Cardiovasc. Pharmacol. 1982, 4, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Vaitkevicius, P.V.; Fleg, J.L.; Engel, J.H.; O’Connor, F.C.; Wright, J.G.; Lakatta, L.E.; Yin, F.C.; Lakatta, E.G. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation 1993, 88, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.F.; Parise, H.; Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Vita, J.A.; Vasan, R.S.; Levy, D. Changes in arterial stiffness and wave reflection with advancing age in healthy men and women: The Framingham heart study. Hypertension 2004, 43, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part iii: Cellular and molecular clues to heart and arterial aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Toda, N. Age-related changes in endothelial function and blood flow regulation. Pharmacol. Ther. 2012, 133, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Matz, R.L.; Andriantsitohaina, R. Age-related endothelial dysfunction: Potential implications for pharmacotherapy. Drugs Aging 2003, 20, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Virdis, A.; Ghiadoni, L.; Giannarelli, C.; Taddei, S. Endothelial dysfunction and vascular disease in later life. Maturitas 2010, 67, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Angulo, J.; Vallejo, S.; El Assar, M.; Garcia-Septiem, J.; Sanchez-Ferrer, C.F.; Rodriguez-Manas, L. Age-related differences in the effects of α and γ peroxisome proliferator-activated receptor subtype agonists on endothelial vasodilation in human microvessels. Exp. Gerontol. 2012, 47, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Mombouli, J.V.; Taylor, A.A.; Vanhoutte, P.M. Endothelium-dependent hyperpolarization caused by bradykinin in human coronary arteries. J. Clin. Investig. 1993, 92, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- Idris Khodja, N.; Chataigneau, T.; Auger, C.; Schini-Kerth, V.B. Grape-derived polyphenols improve aging-related endothelial dysfunction in rat mesenteric artery: Role of oxidative stress and the angiotensin system. PLoS ONE 2012, 7, e32039. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Fujii, K.; Kansui, Y.; Iida, M. Changes in endothelium-derived hyperpolarizing factor in hypertension and ageing: Response to chronic treatment with renin-angiotensin system inhibitors. Clin. Exp. Pharmacol. Physiol. 2004, 31, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-de-Souza, J.L.; Varanda, W.A.; Tostes, R.C.; Chignalia, A.Z. BK channels in cardiovascular diseases and aging. Aging Dis. 2013, 4, 38–49. [Google Scholar] [PubMed]
- Albarwani, S.; Al-Siyabi, S.; Baomar, H.; Hassan, M.O. Exercise training attenuates ageing-induced BKCa channel downregulation in rat coronary arteries. Exp. Physiol. 2010, 95, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Luo, N.; Chi, Y. Aging-shifted prostaglandin profile in endothelium as a factor in cardiovascular disorders. J. Aging Res. 2012, 2012, 121390. [Google Scholar] [CrossRef] [PubMed]
- Schrage, W.G.; Eisenach, J.H.; Joyner, M.J. Ageing reduces nitric-oxide- and prostaglandin-mediated vasodilatation in exercising humans. J. Physiol. 2007, 579, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, W.T.; Vaa, B.; Hesse, C.; Eisenach, J.H.; Joyner, M.J. Aging is associated with reduced prostacyclin-mediated dilation in the human forearm. Hypertension 2009, 53, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Heymes, C.; Habib, A.; Yang, D.; Mathieu, E.; Marotte, F.; Samuel, J.; Boulanger, C.M. Cyclo-oxygenase-1 and -2 contribution to endothelial dysfunction in ageing. Br. J. Pharmacol. 2000, 131, 804–810. [Google Scholar] [CrossRef] [PubMed]
- De Sotomayor, M.A.; Perez-Guerrero, C.; Herrrera, M.D.; Jimenez, L.; Marin, R.; Marhuenda, E.; Andriantsitohaina, R. Improvement of age-related endothelial dysfunction by simvastatin: Effect on no and cox pathways. Br. J. Pharmacol. 2005, 146, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Man, R.Y.; Vanhoutte, P.M. Two isoforms of cyclooxygenase contribute to augmented endothelium-dependent contractions in femoral arteries of 1-year-old rats. Acta Pharmacol. Sin. 2008, 29, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.F.; Huri, D.A.; Snyder, S.H. Inducible nitric oxide synthase binds, s-nitrosylates, and activates cyclooxygenase-2. Science 2005, 310, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase SIRT6 regulates glucose homeostasis via HIF1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, M.; Shimokawa, H.; Otsuji, Y.; Ueta, Y.; Sasaguri, Y.; Yanagihara, N. Nitric oxide synthases and cardiovascular diseases: Insights from genetically modified mice. Circ. J. 2009, 73, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Dere, E.; de Souza Silva, M.A.; Topic, B.; Fiorillo, C.; Li, J.S.; Sadile, A.G.; Frisch, C.; Huston, J.P. Aged endothelial nitric oxide synthase knockout mice exhibit higher mortality concomitant with impaired open-field habituation and alterations in forebrain neurotransmitter levels. Genes Brain Behav. 2002, 1, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Huang, A.; Yan, E.H.; Wu, Z.; Yan, C.; Kaminski, P.M.; Oury, T.D.; Wolin, M.S.; Kaley, G. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2249–H2256. [Google Scholar] [CrossRef] [PubMed]
- Challah, M.; Nadaud, S.; Philippe, M.; Battle, T.; Soubrier, F.; Corman, B.; Michel, J.B. Circulating and cellular markers of endothelial dysfunction with aging in rats. Am. J. Physiol. 1997, 273, H1941–H1948. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Montoya, N.; Giraldo, J.; Vila, E. Ageing affects nitric oxide synthase, cyclooxygenase and oxidative stress enzymes expression differently in mesenteric resistance arteries. Auton. Autacoid Pharmacol. 2005, 25, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Cau, S.B.; Carneiro, F.S.; Tostes, R.C. Differential modulation of nitric oxide synthases in aging: Therapeutic opportunities. Front. Physiol. 2012, 3, 218. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct evidence of endothelial oxidative stress with aging in humans: Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-κB. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Eskurza, I.; Monahan, K.D.; Robinson, J.A.; Seals, D.R. Effect of acute and chronic ascorbic acid on flow-mediated dilatation with sedentary and physically active human ageing. J. Physiol. 2004, 556, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Kimura, M.; Noma, K.; Hara, K.; Jitsuiki, D.; Goto, C.; Oshima, T.; Chayama, K.; et al. Tetrahydrobiopterin improves aging-related impairment of endothelium-dependent vasodilation through increase in nitric oxide production. Atherosclerosis 2006, 186, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Versari, D.; Salvetti, A. Endothelium, aging, and hypertension. Curr. Hypertens. Rep. 2006, 8, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Malinin, N.L.; West, X.Z.; Byzova, T.V. Oxidation as “the stress of life”. Aging (Albany NY) 2011, 3, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Park, W.H. The effects of exogenous H2O2 on cell death, reactive oxygen species and glutathione levels in calf pulmonary artery and human umbilical vein endothelial cells. Int. J. Mol. Med. 2013, 31, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, P.; Popp, R.; Wiegand, B.; Altschmied, J.; Haendeler, J. Nuclear redox-signaling is essential for apoptosis inhibition in endothelial cells—important role for nuclear thioredoxin-1. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Groleau, J.; Dussault, S.; Turgeon, J.; Haddad, P.; Rivard, A. Accelerated vascular aging in cuznsod-deficient mice: Impact on epc function and reparative neovascularization. PLoS ONE 2011, 6, e23308. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, J.; Haddad, P.; Dussault, S.; Groleau, J.; Maingrette, F.; Perez, G.; Rivard, A. Protection against vascular aging in NOX2-deficient mice: Impact on endothelial progenitor cells and reparative neovascularization. Atherosclerosis 2012, 223, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Wolin, M.S. Reactive oxygen species and the control of vascular function. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H539–H549. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Lassegue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Fridovich, I. Mitochondria: Are they the seat of senescence? Aging Cell 2004, 3, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, B.; Schildknecht, S.; Zee, R.; Bachschmid, M.M. Signalling processes in endothelial ageing in relation to chronic oxidative stress and their potential therapeutic implications in humans. Exp. Physiol. 2009, 94, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Sonntag, W.E.; Csiszar, A. Mitochondria and aging in the vascular system. J. Mol. Med. 2010, 88, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.H.; Vendrov, A.E.; Tchivilev, I.; Niu, X.L.; Molnar, K.C.; Rojas, M.; Carter, J.D.; Tong, H.; Stouffer, G.A.; Madamanchi, N.R.; et al. Mitochondrial oxidative stress in aortic stiffening with age: The role of smooth muscle cell function. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Bovina, C.; Castelluccio, C.; Fato, R.; Formiggini, G.; Genova, M.L.; Marchetti, M.; Pich, M.M.; Pallotti, F.; Parenti Castelli, G.; et al. Mitochondrial complex I defects in aging. Mol. Cell. Biochem. 1997, 174, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Gutterman, D.D. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2023–H2031. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.H. Aging: A revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp. Gerontol. 2007, 42, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of NOX1 and NOX4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; O’Donnell, V.B.; Wood, J.D.; Broughton, J.P.; Hughes, E.J.; Jones, O.T. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am. J. Physiol. 1996, 271, H1626–H1634. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin ii. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.; Harrison, D.G.; Griendling, K.K. Distinct roles of NOX1 and NOX4 in basal and angiotensin ii-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. NOX4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase NOX4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [PubMed]
- Bode-Boger, S.M.; Muke, J.; Surdacki, A.; Brabant, G.; Boger, R.H.; Frolich, J.C. Oral l-arginine improves endothelial function in healthy individuals older than 70 years. Vasc. Med. 2003, 8, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, L.; Christianson, D.W.; Nyhan, D.; Berkowitz, D.E. Arginase and vascular aging. J. Appl. Physiol. 2008, 105, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bugaj, L.J.; Oh, Y.J.; Bivalacqua, T.J.; Ryoo, S.; Soucy, K.G.; Santhanam, L.; Webb, A.; Camara, A.; Sikka, G.; et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J. Appl. Physiol. 2009, 107, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.D.; Quinlan, C.L.; Andrukhiv, A.; West, I.C.; Jaburek, M.; Garlid, K.D. The direct physiological effects of mitok(ATP) opening on heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H406–H415. [Google Scholar] [CrossRef] [PubMed]
- Queliconi, B.B.; Wojtovich, A.P.; Nadtochiy, S.M.; Kowaltowski, A.J.; Brookes, P.S. Redox regulation of the mitochondrial k(ATP) channel in cardioprotection. Biochim. Biophys. Acta 2011, 1813, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin ii-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, A.; Orru, M.; Morrell, C.; Piras, M.G.; Taub, D.; Schlessinger, D.; Uda, M.; Lakatta, E.G. Independent and additive effects of cytokine patterns and the metabolic syndrome on arterial aging in the SardiNIA study. Atherosclerosis 2011, 215, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Miles, E.A.; Rees, D.; Banerjee, T.; Cazzola, R.; Lewis, S.; Wood, R.; Oates, R.; Tallant, A.; Cestaro, B.; Yaqoob, P.; et al. Age-related increases in circulating inflammatory markers in men are independent of bmi, blood pressure and blood lipid concentrations. Atherosclerosis 2008, 196, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, L.; Chong, K.; Franco, R.; Rosati, A.; de Caro, F.; Capunzo, M.; Turco, M.C.; Hoon, D.S. BAG3 protein expression in melanoma metastatic lymph nodes correlates with patients’ survival. Cell Death Dis. 2014, 5, e1173. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Kaley, G. Vascular inflammation in aging. Herz 2004, 29, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Mattace-Raso, F.U.; van der Cammen, T.J.; van der Meer, I.M.; Schalekamp, M.A.; Asmar, R.; Hofman, A.; Witteman, J.C. C-reactive protein and arterial stiffness in older adults: The rotterdam study. Atherosclerosis 2004, 176, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Nakhai-Pour, H.R.; Grobbee, D.E.; Bots, M.L.; Muller, M.; van der Schouw, Y.T. C-reactive protein and aortic stiffness and wave reflection in middle-aged and elderly men from the community. J. Hum. Hypertens. 2007, 21, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, J.; Jiang, L.Q.; Spinetti, G.; Pintus, G.; Monticone, R.; Kolodgie, F.D.; Virmani, R.; Lakatta, E.G. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 2007, 50, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Ungvari, Z.; Koller, A.; Edwards, J.G.; Kaley, G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genom. 2004, 17, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Vila, E.; Salaices, M. Cytokines and vascular reactivity in resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1016–H1021. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Edwards, J.G.; Kaminski, P.M.; Wolin, M.S.; Kaley, G.; Koller, A. Increased superoxide production in coronary arteries in hyperhomocysteinemia: Role of tumor necrosis factor-α, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Nevado, J.; Vallejo, S.; El-Assar, M.; Peiro, C.; Sanchez-Ferrer, C.F.; Rodriguez-Manas, L. Changes in the human peritoneal mesothelial cells during aging. Kidney Int. 2006, 69, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Mountain, D.J.; Singh, M.; Menon, B.; Singh, K. Interleukin-1β increases expression and activity of matrix metalloproteinase-2 in cardiac microvascular endothelial cells: Role of PKCα/β1 and MAPKs. Am. J. Physiol. Cell Physiol. 2007, 292, C867–875. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Lee, S.H.; Park, D.W.; Bae, Y.S.; Yun, S.S.; Kim, J.R.; Baek, S.H. Phosphatidic acid as a regulator of matrix metalloproteinase-9 expression via the TNF-α signaling pathway. FEBS Lett. 2007, 581, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Spinetti, G.; Monticone, R.E.; Zhang, J.; Wu, J.; Jiang, L.; Khazan, B.; Telljohann, R.; Lakatta, E.G. A local proinflammatory signalling loop facilitates adverse age-associated arterial remodeling. PLoS ONE 2011, 6, e16653. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Jat, P.S. Deciphering the role of nuclear factor-κB in cellular senescence. Aging (Albany NY) 2011, 3, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Sinha, S.; Kawahara, T.L.; Zhang, J.Y.; Segal, E.; Chang, H.Y. Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev. 2007, 21, 3244–3257. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.P.; Chung, H.Y. Adaptive mechanisms to oxidative stress during aging. Mech. Ageing Dev. 2006, 127, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Orosz, Z.; Labinskyy, N.; Rivera, A.; Xiangmin, Z.; Smith, K.; Csiszar, A. Increased mitochondrial H2O2 production promotes endothelial NF-κB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H37–H47. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.L.; Lesniewski, L.A.; Lawson, B.R.; Beske, S.D.; Seals, D.R. Nuclear factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation 2009, 119, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Takagi, G.; Asai, K.; Resuello, R.G.; Natividad, F.F.; Vatner, D.E.; Vatner, S.F.; Lakatta, E.G. Aging increases aortic MMP-2 activity and angiotensin ii in nonhuman primates. Hypertension 2003, 41, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Monticone, R.E.; Lakatta, E.G. Arterial aging: A journey into subclinical arterial disease. Curr. Opin. Nephrol. Hypertens. 2010, 19, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, J.; Walker, S.J.; Dworakowski, R.; Lakatta, E.G.; Shah, A.M. Involvement of NADPH oxidase in age-associated cardiac remodeling. J. Mol. Cell. Cardiol. 2010, 48, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.D.; Frackowiak, R.S. Cerebral blood flow and metabolism studies in multi-infarct dementia. Alzheimer Dis. Assoc. Disord. 1991, 5, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Strandgaard, S.; Olesen, J.; Skinhoj, E.; Lassen, N.A. Autoregulation of brain circulation in severe arterial hypertension. Br. Med. J. 1973, 1, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Keady, J.; Swarbrick, C.; Deaton, C.; Reilly, S.; Price, R.; Howorth, M.; Riley, C.; Lowndes, J.; Pendleton, N. Proposing a cardiac model for vascular dementia. Br. J. Nurs. 2012, 21, 1124. [Google Scholar] [PubMed]
- Elahy, M.; Jackaman, C.; Mamo, J.C.; Lam, V.; Dhaliwal, S.S.; Giles, C.; Nelson, D.; Takechi, R. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun. Ageing 2015, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D. Effect of aging on the blood-brain barrier. Neurobiol. Aging 1988, 9, 31–39. [Google Scholar] [CrossRef]
- Mooradian, A.D. Potential mechanisms of the age-related changes in the blood-brain barrier. Neurobiol. Aging 1994, 15, 751–755, discussion 761–752, 767. [Google Scholar] [CrossRef]
- Wikkelso, C.; Blomstrand, C.; Nordquist, P. Cerebrospinal fluid investigations in multi-infarct dementia and senile dementia. Acta Neurol. Scand. 1981, 64, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Strandgaard, S. The cerebral circulation in the elderly: The influence of age, vascular disease, and antihypertensive treatment. Am. J. Geriatr. Cardiol. 1993, 2, 32–36. [Google Scholar] [PubMed]
- Sulkava, R.; Erkinjuntti, T. Vascular dementia due to cardiac arrhythmias and systemic hypotension. Acta Neurol. Scand. 1987, 76, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S.; Rogers, R.L.; Mortel, K.F. Progressive cerebral ischemia antedates cerebrovascular symptoms by two years. Ann. Neurol. 1984, 16, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, G.; Tweedy, J.R. Cerebral blood flow in severity-matched Alzheimer and multi-infarct patients. Neurology 1987, 37, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Deary, I.J.; Corley, J.; Gow, A.J.; Harris, S.E.; Houlihan, L.M.; Marioni, R.E.; Penke, L.; Rafnsson, S.B.; Starr, J.M. Age-associated cognitive decline. Br. Med. Bull. 2009, 92, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Hossmann, K.A. The two pathophysiologies of focal brain ischemia: Implications for translational stroke research. J. Cereb. Blood Flow Metab. 2012, 32, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Schaller, B.J. Influence of age on stroke and preconditioning-induced ischemic tolerance in the brain. Exp. Neurol. 2007, 205, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.P., Jr.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E., 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. Toast. Trial of org 10172 in acute stroke treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kissela, B.M.; Khoury, J.C.; Alwell, K.; Moomaw, C.J.; Woo, D.; Adeoye, O.; Flaherty, M.L.; Khatri, P.; Ferioli, S.; de Los Rios La Rosa, F.; et al. Age at stroke: Temporal trends in stroke incidence in a large, biracial population. Neurology 2012, 79, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Carmichael, S.T.; Kokaia, Z.; Kessler, C.; Walker, L.C. The response of the aged brain to stroke: Too much, too soon? Curr. Neurovasc. Res. 2007, 4, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Chollet, F. Pharmacologic approaches to cerebral aging and neuroplasticity: Insights from the stroke model. Dialogues Clin. Neurosci. 2013, 15, 67–76. [Google Scholar] [PubMed]
- Pasqualin, A. Epidemiology and pathophysiology of cerebral vasospasm following subarachnoid hemorrhage. J. Neurosurg. Sci. 1998, 42, 15–21. [Google Scholar] [PubMed]
- Mayberg, M. Pathophysiology, monitoring, and treatment of cerebral vasospasm after subarachnoid hemorrhage. J. Stroke Cerebrovasc. Dis. 1997, 6, 258–260. [Google Scholar] [CrossRef]
- Gathier, C.S.; Dankbaar, J.W.; van der Jagt, M.; Verweij, B.H.; Oldenbeuving, A.W.; Rinkel, G.J.; van den Bergh, W.M.; Slooter, A.J.; Group, H.S. Effects of induced hypertension on cerebral perfusion in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: A randomized clinical trial. Stroke 2015, 46, 3277–3281. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.K.; Kapadia, A.; Ilodigwe, D.; Li, Z.; Schweizer, T.A.; Macdonald, R.L. Impact of global cerebral atrophy on clinical outcome after subarachnoid hemorrhage. J. Neurosurg. 2013, 119, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.F.; Song, Y.F.; Liu, H.B.; Liu, Z.; Wang, S.S. The clinical characteristics and treatment of cerebral microarteriovenous malformation presenting with intracerebral hemorrhage: A series of 13 cases. BioMed Res. Int. 2015, 2015, 257153. [Google Scholar] [CrossRef] [PubMed]
- Gold, G.; Giannakopoulos, P.; Montes-Paixao Junior, C.; Herrmann, F.R.; Mulligan, R.; Michel, J.P.; Bouras, C. Sensitivity and specificity of newly proposed clinical criteria for possible vascular dementia. Neurology 1997, 49, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016, 131, 659–685. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Tsukagoshi, H.; Otomo, E.; Hayakawa, M. Cerebral amyloid angiopathy in the aged. J. Neurol. 1987, 234, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Akiguchi, I. Pathophysiology and therapeutic approaches on Binswanger’s disease. No To Shinkei 2006, 58, 289–297. [Google Scholar] [PubMed]
- Merkli, H.; Pal, E.; Horvathne, V.I. Clinical characteristics of subcortical arteriosclerotic encephalopathy (Binswanger’s disease). Orv. Hetil. 2001, 142, 1221–1226. [Google Scholar] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; Forte, M.; Corbi, G.; Russomanno, G.; Formisano, L.; Landolfi, A.; Izzo, V.; Filippelli, A.; Vecchione, C.; Carrizzo, A. Sirtuins: Possible clinical implications in cardio and cerebrovascular diseases. Curr. Drug Targets 2017, 18, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Qadir, M.I.; Anwar, S. Sirtuins in brain aging and neurological disorders. Crit. Rev. Eukaryot. Gene Expr. 2017, 27, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, W.; Liu, Y.; Sun, Y.; Li, Y.; Yao, Q.; Li, J.; Zhang, Q.; Gao, Y.; Gao, L.; et al. α-lipoic acid improves high-fat diet-induced hepatic steatosis by modulating the transcription factors SREBP-1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J. Nutr. Biochem. 2014, 25, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, W.; McBurney, M.W.; Longo, V.D. SIRT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008, 8, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Burnett, C.; Valentini, S.; Cabreiro, F.; Goss, M.; Somogyvari, M.; Piper, M.D.; Hoddinott, M.; Sutphin, G.L.; Leko, V.; McElwee, J.J.; et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 2011, 477, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Inuzuka, Y.; Okuda, J.; Kato, T.; Niizuma, S.; Tamaki, Y.; Iwanaga, Y.; Kawamoto, A.; Narazaki, M.; Matsuda, T.; et al. Constitutive SIRT1 overexpression impairs mitochondria and reduces cardiac function in mice. J. Mol. Cell. Cardiol. 2011, 51, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. SIRT1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gao, P.; Zhang, H.; Chen, H.; Zheng, W.; Lv, X.; Xu, T.; Wei, Y.; Liu, D.; Liang, C. SIRT1 inhibits angiotensin II-induced vascular smooth muscle cell hypertrophy. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The deacetylase SIRT1 promotes membrane localization and activation of AKT and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef] [PubMed]
- Takemura, A.; Iijima, K.; Ota, H.; Son, B.K.; Ito, Y.; Ogawa, S.; Eto, M.; Akishita, M.; Ouchi, Y. Sirtuin 1 retards hyperphosphatemia-induced calcification of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. SIRT3 blocks the cardiac hypertrophic response by augmenting FOXO3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Yang, X.; Liu, T.; Zhang, T.; Xie, Q.R.; Xia, W. Autophagy induction by SIRT6 is involved in oxidative stress-induced neuronal damage. Protein Cell 2016, 7, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. SIRT7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Le Roith, D. Seminars in medicine of the beth israel deaconess medical center. Insulin-like growth factors. N. Engl. J. Med. 1997, 336, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Guttridge, D.C. Signaling pathways weigh in on decisions to make or break skeletal muscle. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Mancino, M.G.; Onori, P.; Franchitto, A.; Alpini, G.; Francis, H.; Glaser, S.; Gaudio, E. Estrogens and the pathophysiology of the biliary tree. World J. Gastroenterol. 2006, 12, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase AKT. Science 1997, 275, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor 1 (IGF-1) and type 1 IGF receptor (IGF1R). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Ruvinov, E.; Leor, J.; Cohen, S. The promotion of myocardial repair by the sequential delivery of IGF-1 and HGF from an injectable alginate biomaterial in a model of acute myocardial infarction. Biomaterials 2011, 32, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Delaughter, M.C.; Taffet, G.E.; Fiorotto, M.L.; Entman, M.L.; Schwartz, R.J. Local insulin-like growth factor i expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. FASEB J. 1999, 13, 1923–1929. [Google Scholar] [CrossRef] [PubMed]
- Frater, J.; Lie, D.; Bartlett, P.; McGrath, J.J. Insulin-like growth factor 1 (IGF-1) as a marker of cognitive decline in normal ageing: A review. Ageing Res. Rev. 2017, 42, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Arden, K.C. FOXO animal models reveal a variety of diverse roles for FOXO transcription factors. Oncogene 2008, 27, 2345–2350. [Google Scholar] [CrossRef] [PubMed]
- Sedding, D.G. FOXO transcription factors in oxidative stress response and ageing—A new fork on the way to longevity? Biol. Chem. 2008, 389, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Kitayama, K.; Shimoda, Y.; Ogawa, M.; Sone, K.; Yoshida-Araki, K.; Hisatsune, H.; Nishikawa, S.; Nakayama, K.; Nakayama, K.; et al. Abnormal angiogenesis in FOXO1 (FKHR)-deficient mice. J. Biol. Chem. 2004, 279, 34741–34749. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Urbich, C.; Sasaki, K.; Hofmann, W.K.; Heeschen, C.; Aicher, A.; Kollipara, R.; DePinho, R.A.; Zeiher, A.M.; Dimmeler, S. Involvement of foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Investig. 2005, 115, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wang, G.; Peng, J.; Qian, G.; Jiang, W.; Xie, C.; Xiao, Y.; Wang, X. Knockdown of SIRT1 suppresses bladder cancer cell proliferation and migration and induces cell cycle arrest and antioxidant response through FOXO3a-mediated pathways. Biomed Res. Int. 2017, 2017, 3781904. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived MCLK1+/− mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Yee, C.; Wang, Y.; Hekimi, S. A single biochemical activity underlies the pleiotropy of the aging-related protein CLK-1. Sci. Rep. 2017, 7, 859. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Boutis, P.; Hekimi, S. Mutations in the CLK-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 1995, 139, 1247–1259. [Google Scholar] [PubMed]
- Levavasseur, F.; Miyadera, H.; Sirois, J.; Tremblay, M.L.; Kita, K.; Shoubridge, E.; Hekimi, S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J. Biol. Chem. 2001, 276, 46160–46164. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Lapointe, J.; Hekimi, S. Lifelong protection from global cerebral ischemia and reperfusion in long-lived MCLK1+/− mutants. Exp. Neurol. 2010, 223, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Giorgio, M.; Ferdinandy, P.; Schulz, R. New aspects of p66shc in ischaemia reperfusion injury and other cardiovascular diseases. Br. J. Pharmacol. 2017, 174, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Betts, D.H.; Bain, N.T.; Madan, P. The p66(shc) adaptor protein controls oxidative stress response in early bovine embryos. PLoS ONE 2014, 9, e86978. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Francia, P.; Camici, G.G.; Pelicci, P.G.; Luscher, T.F.; Volpe, M. Final common molecular pathways of aging and cardiovascular disease: Role of the p66shc protein. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Martin-Padura, I.; de Nigris, F.; Giorgio, M.; Mansueto, G.; Somma, P.; Condorelli, M.; Sica, G.; de Rosa, G.; Pelicci, P. Deletion of the p66shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc. Natl. Acad. Sci. USA 2003, 100, 2112–2116. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yu, Y.; Montani, J.P.; Yang, Z.; Ming, X.F. Arginase-II induces vascular smooth muscle cell senescence and apoptosis through p66shc and p53 independently of its l-arginine ureahydrolase activity: Implications for atherosclerotic plaque vulnerability. J. Am. Heart Assoc. 2013, 2, e000096. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Klotho. Pflugers Arch. 2010, 459, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Dalton, G.D.; Xie, J.; An, S.W.; Huang, C.L. New insights into the mechanism of action of soluble klotho. Front. Endocrinol. (Lausanne) 2017, 8, 323. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, L.; Yang, J.; Hao, L. Activation of peroxisome proliferator-activated receptor gamma inhibits vascular calcification by upregulating Klotho. Exp. Ther. Med. 2017, 13, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Maltese, G.; Fountoulakis, N.; Siow, R.C.; Gnudi, L.; Karalliedde, J. Perturbations of the anti-ageing hormone Klotho in patients with type 1 diabetes and microalbuminuria. Diabetologia 2017, 60, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Pasini, E.; Scarabelli, T.M.; Romano, C.; Agrawal, P.R.; Chen-Scarabelli, C.; Knight, R.; Saravolatz, L.; Narula, J.; Ferrari-Vivaldi, M.; et al. Decreased expression of Klotho in cardiac atria biopsy samples from patients at higher risk of atherosclerotic cardiovascular disease. J. Geriatr. Cardiol. 2016, 13, 701–711. [Google Scholar] [PubMed]
- Takeshita, K.; Fujimori, T.; Kurotaki, Y.; Honjo, H.; Tsujikawa, H.; Yasui, K.; Lee, J.K.; Kamiya, K.; Kitaichi, K.; Yamamoto, K.; et al. Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation 2004, 109, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Bolanos-Garcia, V.M.; Blundell, T.L. BUB1 and BubR1: Multifaceted kinases of the cell cycle. Trends Biochem. Sci. 2011, 36, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jeganathan, K.B.; Malureanu, L.; Perez-Terzic, C.; Terzic, A.; van Deursen, J.M. Early aging-associated phenotypes in BUB3/RAE1 haploinsufficient mice. J. Cell Biol. 2006, 172, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Baker, D.J.; d’Uscio, L.V.; Mozammel, G.; Katusic, Z.S.; van Deursen, J.M. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke 2007, 38, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Okadome, J.; Matsumoto, T.; Yoshiya, K.; Matsuda, D.; Tamada, K.; Onimaru, M.; Nakano, K.; Egashira, K.; Yonemitsu, Y.; Maehara, Y. BubR1 insufficiency impairs angiogenesis in aging and in experimental critical limb ischemic mice. J. Vasc. Surg. 2017. [Google Scholar] [CrossRef] [PubMed]
- Guntani, A.; Matsumoto, T.; Kyuragi, R.; Iwasa, K.; Onohara, T.; Itoh, H.; Katusic, Z.S.; Maehara, Y. Reduced proliferation of aged human vascular smooth muscle cells–role of oxygen-derived free radicals and BubR1 expression. J. Surg. Res. 2011, 170, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.M.Q.; Jang, M.H. BubR1 and brain aging. Aging (Albany NY) 2017, 9, 1955–1956. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, C.V.; Malovini, A.; Roncarati, R.; Novelli, V.; Villa, F.; Condorelli, G.; Bellazzi, R.; Puca, A.A. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009, 12, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Nebel, A.; Kleindorp, R.; Caliebe, A.; Nothnagel, M.; Blanche, H.; Junge, O.; Wittig, M.; Ellinghaus, D.; Flachsbart, F.; Wichmann, H.E.; et al. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech. Ageing Dev. 2011, 132, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Geesaman, B.J.; Benson, E.; Brewster, S.J.; Kunkel, L.M.; Blanche, H.; Thomas, G.; Perls, T.T.; Daly, M.J.; Puca, A.A. Haplotype-based identification of a microsomal transfer protein marker associated with the human lifespan. Proc. Natl. Acad. Sci. USA 2003, 100, 14115–14120. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Carrizzo, A.; Spinelli, C.C.; Ferrario, A.; Malovini, A.; Maciag, A.; Damato, A.; Auricchio, A.; Spinetti, G.; Sangalli, E.; et al. Genetic analysis reveals a longevity-associated protein modulating endothelial function and angiogenesis. Circ. Res. 2015, 117, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, C.C.; Carrizzo, A.; Ferrario, A.; Villa, F.; Damato, A.; Ambrosio, M.; Madonna, M.; Frati, G.; Fucile, S.; Sciaccaluga, M.; et al. LAV-BPIFB4 isoform modulates eNOS signalling through Ca2+/PKC-α-dependent mechanism. Cardiovasc. Res. 2017, 113, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Malovini, A.; Carrizzo, A.; Spinelli, C.C.; Ferrario, A.; Maciag, A.; Madonna, M.; Bellazzi, R.; Milanesi, L.; Vecchione, C.; et al. Serum BPIFB4 levels classify health status in long-living individuals. Immun. Ageing 2015, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Belcher, M.; van der Harst, P. Healthy aging and disease: Role for telomere biology? Clin. Sci. (Lond.) 2011, 120, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Edo, M.D.; Andres, V. Aging, telomeres, and atherosclerosis. Cardiovasc. Res. 2005, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fyhrquist, F.; Saijonmaa, O.; Strandberg, T. The roles of senescence and telomere shortening in cardiovascular disease. Nat. Rev. Cardiol. 2013, 10, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J. Stem cells and regeneration of the cardiovascular system: Facts, fictions, and uncertainties. Blood Cells Mol. Dis. 2004, 32, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Ballard, V.L.; Edelberg, J.M. Stem cells and the regeneration of the aging cardiovascular system. Circ. Res. 2007, 100, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Csobonyeiova, M.; Polak, S.; Danisovic, L. Perspectives of induced pluripotent stem cells for cardiovascular system regeneration. Exp. Biol. Med. (Maywood) 2015, 240, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, Z.; Sun, Z. The potential and challenges of using stem cells for cardiovascular repair and regeneration. Genes Dis. 2014, 1, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Fusco, D.; Colloca, G.; Lo Monaco, M.R.; Cesari, M. Effects of antioxidant supplementation on the aging process. Clin. Interv. Aging 2007, 2, 377–387. [Google Scholar] [PubMed]
- Kirkland, J.L. The biology of senescence: Potential for prevention of disease. Clin. Geriatr. Med. 2002, 18, 383–405. [Google Scholar] [CrossRef]
- Walston, J.; Hadley, E.C.; Ferrucci, L.; Guralnik, J.M.; Newman, A.B.; Studenski, S.A.; Ershler, W.B.; Harris, T.; Fried, L.P. Research agenda for frailty in older adults: Toward a better understanding of physiology and etiology: Summary from the American geriatrics society/national institute on aging research conference on frailty in older adults. J. Am. Geriatr. Soc. 2006, 54, 991–1001. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izzo, C.; Carrizzo, A.; Alfano, A.; Virtuoso, N.; Capunzo, M.; Calabrese, M.; De Simone, E.; Sciarretta, S.; Frati, G.; Oliveti, M.; et al. The Impact of Aging on Cardio and Cerebrovascular Diseases. Int. J. Mol. Sci. 2018, 19, 481. https://doi.org/10.3390/ijms19020481
Izzo C, Carrizzo A, Alfano A, Virtuoso N, Capunzo M, Calabrese M, De Simone E, Sciarretta S, Frati G, Oliveti M, et al. The Impact of Aging on Cardio and Cerebrovascular Diseases. International Journal of Molecular Sciences. 2018; 19(2):481. https://doi.org/10.3390/ijms19020481
Chicago/Turabian StyleIzzo, Carmine, Albino Carrizzo, Antonia Alfano, Nicola Virtuoso, Mario Capunzo, Mariaconsiglia Calabrese, Eros De Simone, Sebastiano Sciarretta, Giacomo Frati, Marco Oliveti, and et al. 2018. "The Impact of Aging on Cardio and Cerebrovascular Diseases" International Journal of Molecular Sciences 19, no. 2: 481. https://doi.org/10.3390/ijms19020481
APA StyleIzzo, C., Carrizzo, A., Alfano, A., Virtuoso, N., Capunzo, M., Calabrese, M., De Simone, E., Sciarretta, S., Frati, G., Oliveti, M., Damato, A., Ambrosio, M., De Caro, F., Remondelli, P., & Vecchione, C. (2018). The Impact of Aging on Cardio and Cerebrovascular Diseases. International Journal of Molecular Sciences, 19(2), 481. https://doi.org/10.3390/ijms19020481