Fundamental Molecules and Mechanisms for Forming and Maintaining Neuromuscular Synapses


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Rock Bottom Synaptic Transmission
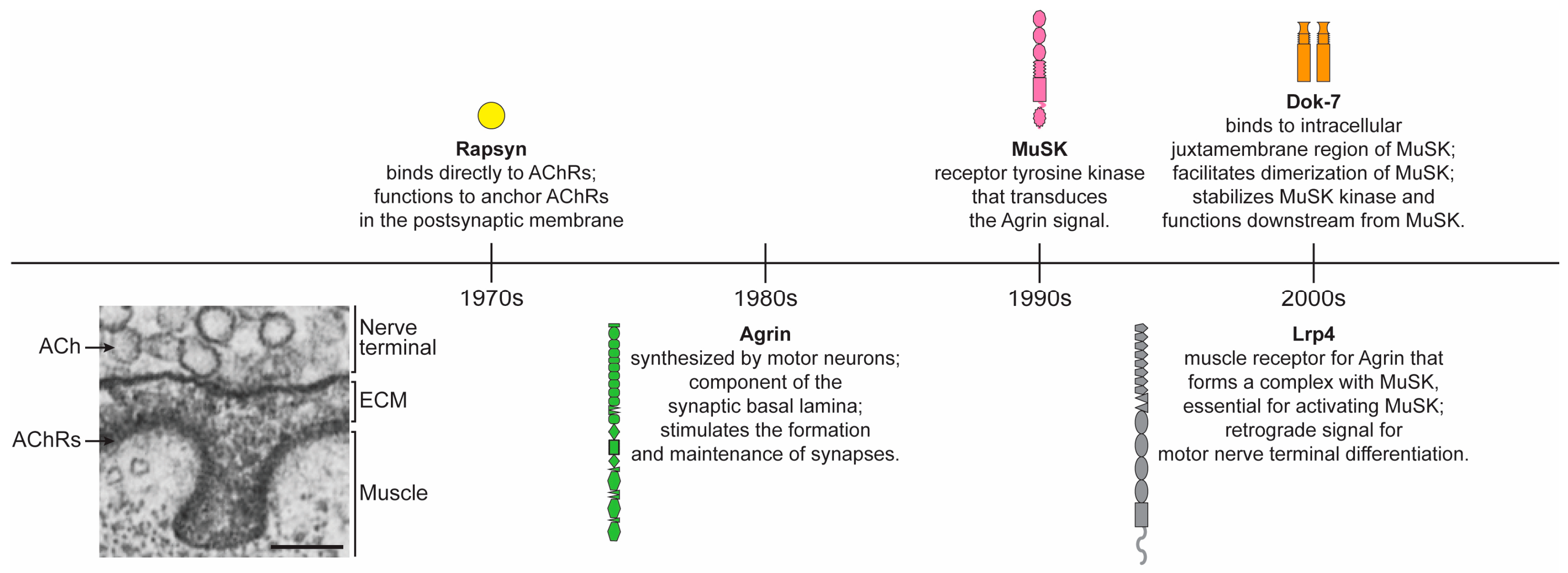
2. Fundamental Proteins of the Neuromuscular Synapse
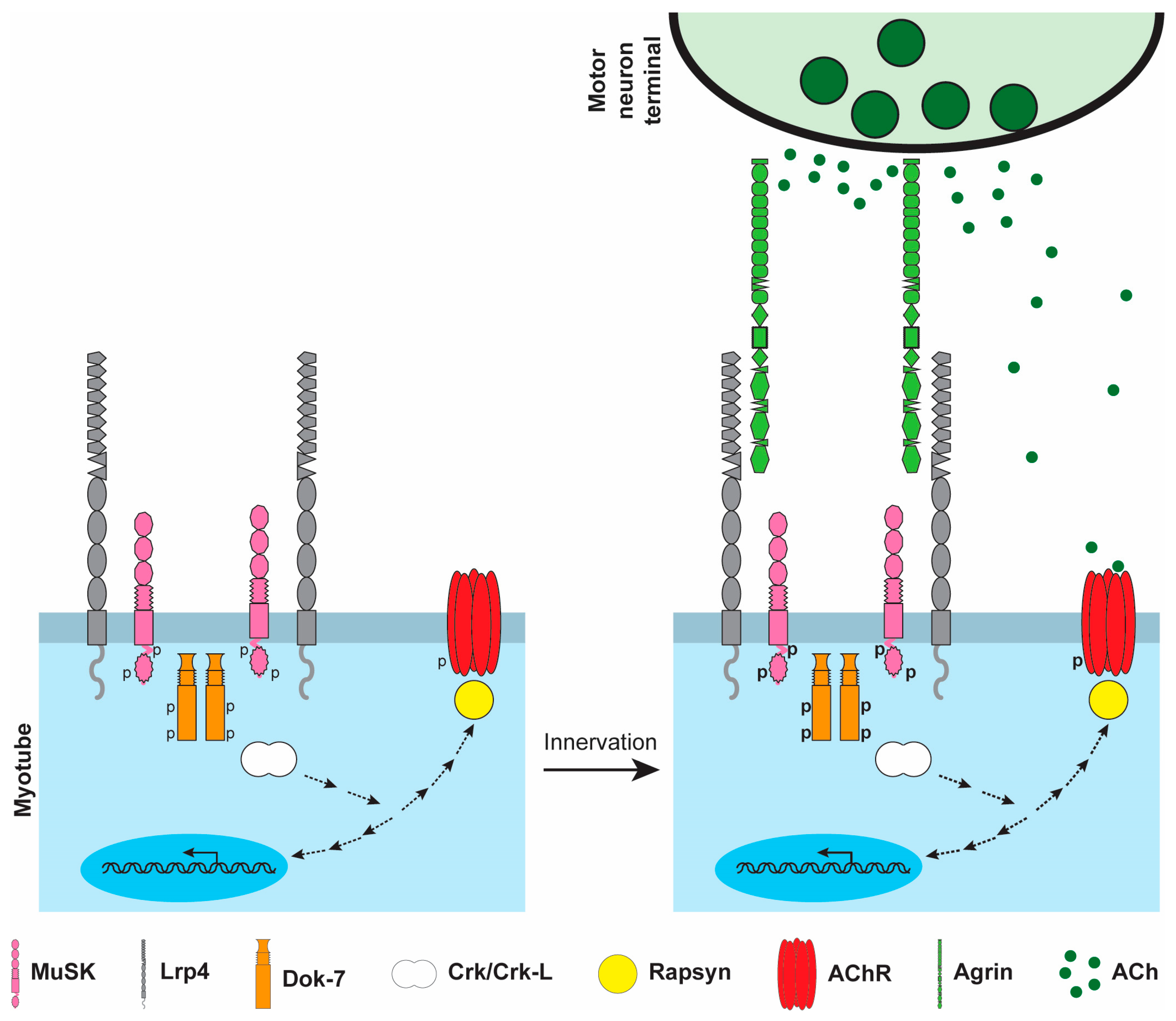
2.1. Agrin
2.2. MuSK
2.3. Lrp4
2.4. Dok-7
2.5. Rapsyn
3. Steps in Synapse Formation
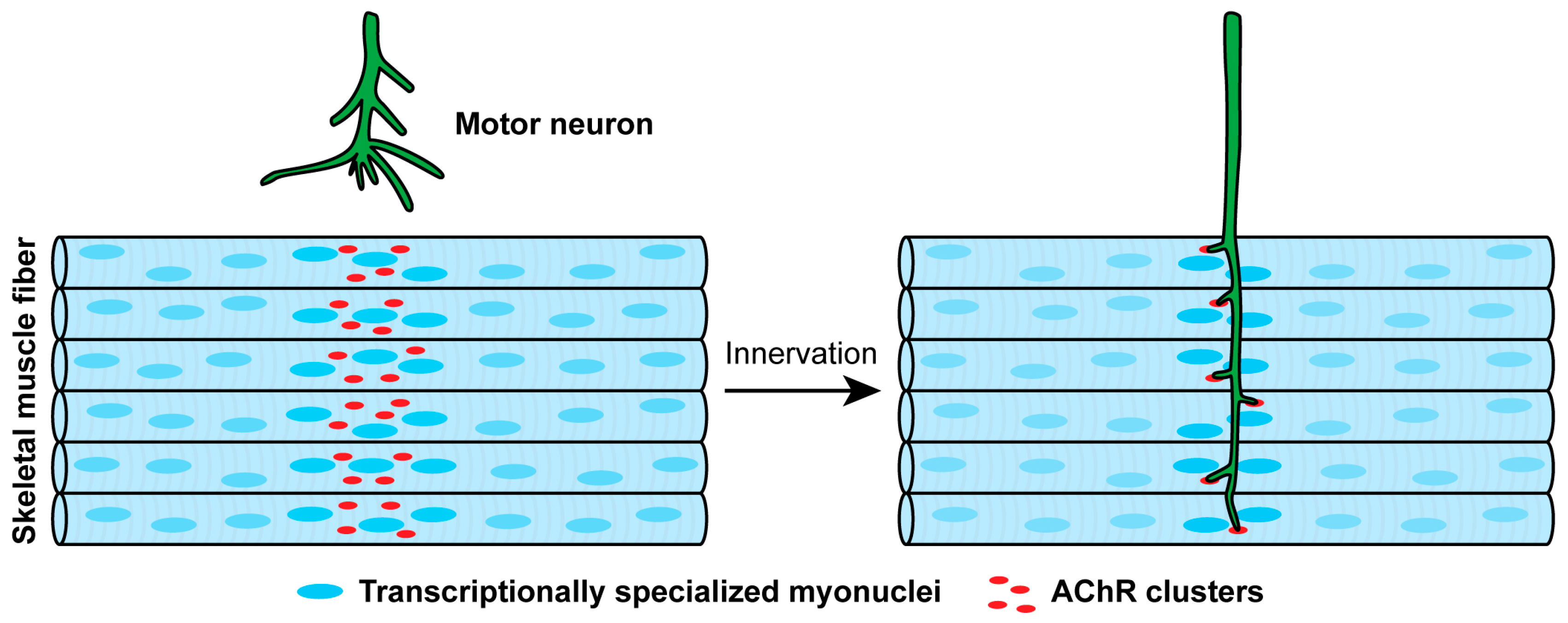
3.1. Muscle Prepatterning
3.2. Stabilizing and Dispersing AChR Clusters
3.3. Synapse Elimination and Maturation
3.4. Targets for Therapy to Maintain Neuromuscular Synapses and Reduce Denervation
4. Conclusions and Discussion
Acknowledgments
Conflicts of Interest
References
- Birks, R.; Katz, B.; Miledi, R. Physiological and structural changes at the amphibian myoneural junction, in the course of nerve degeneration. J. Physiol. 1960, 150, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, R.W.; Lentz, T.L.; Cohen, M.W. Development of the myotomal neuromuscular junction in Xenopus laevis: An electrophysiological and fine-structural study. Dev. Biol. 1977, 60, 101–129. [Google Scholar] [CrossRef]
- McMahan, U.J.; Sanes, J.R.; Marshall, L.M. Cholinesterase is associated with the basal lamina at the neuromuscular junction. Nature 1978, 271, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.M.; Sanes, J.R.; McMahan, U.J. Reinnervation of original synaptic sites on muscle fiber basement membrane after disruption of the muscle cells. Proc. Natl. Acad. Sci. USA 1977, 74, 3073–3077. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R.; Marshall, L.M.; McMahan, U.J. Reinnervation of muscle fiber basal lamina after removal of myofibers. Differentiation of regenerating axons at original synaptic sites. J. Cell Biol. 1978, 78, 176–198. [Google Scholar] [CrossRef] [PubMed]
- Burden, S.J.; Sargent, P.B.; McMahan, U.J. Acetylcholine receptors in regenerating muscle accumulate at original synaptic sites in the absence of the nerve. J. Cell Biol. 1979, 82, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, E.W.; Nitkin, R.M.; Wallace, B.G.; Rubin, L.L.; McMahan, U.J. Components of Torpedo electric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. J. Cell Biol. 1984, 99, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, E.W.; Nitkin, R.M.; Wallace, B.G.; Rubin, L.L.; McMahan, U.J. Identification of Agrin, a synaptic organizing protein from Torpedo electric organ. J. Cell Biol. 1987, 105, 2471–2478. [Google Scholar]
- Magill-Solc, C.; McMahan, U.J. Motor neurons contain agrin-like molecules. J. Cell Biol. 1988, 107, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Noakes, P.G.; Moscoso, L.; Rupp, F.; Scheller, R.H.; Merlie, J.P.; Sanes, J.R. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 1996, 85, 525–535. [Google Scholar] [CrossRef]
- Burgess, R.W.; Nguyen, Q.T.; Son, Y.J.; Lichtman, J.W.; Sanes, J.R. Alternatively spliced isoforms of nerve- and muscle-derived agrin: Their roles at the neuromuscular junction. Neuron 1999, 23, 33–44. [Google Scholar] [CrossRef]
- Bowe, M.A.; Fallon, J.R. The role of agrin in synapse formation. Annu. Rev. Neurosci. 1995, 18, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Denzer, A.J.; Hauser, D.M.; Gesemann, M.; Ruegg, M.A. Synaptic differentiation: The role of agrin in the formation and maintenance of the neuromuscular junction. Cell Tissue Res. 1997, 290, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R.; Lichtman, J.W. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2001, 2, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Hoch, W.; Campanelli, J.T.; Harrison, S.; Scheller, R.H. Structural domains of agrin required for clustering of nicotinic acetylcholine receptors. EMBO J. 1994, 13, 2814–2821. [Google Scholar] [PubMed]
- Burgess, R.W.; Skarnes, W.C.; Sanes, J.R. Agrin isoforms with distinct amino termini: Differential expression, localization, and function. J. Cell Biol. 2000, 151, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Campanelli, J.T.; Roberds, S.L.; Campbell, K.P.; Scheller, R.H. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell 1994, 77, 663–674. [Google Scholar] [CrossRef]
- Gee, S.H.; Montanaro, F.; Lindenbaum, M.H.; Carbonetto, S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell 1994, 77, 675–686. [Google Scholar] [CrossRef]
- Bowe, M.A.; Deyst, K.A.; Leszyk, J.D.; Fallon, J.R. Identification and purification of an agrin receptor from Torpedo postsynaptic membranes: A heteromeric complex related to the dystroglycans. Neuron 1994, 12, 1173–1180. [Google Scholar] [CrossRef]
- Sugiyama, J.; Bowen, D.C.; Hall, Z.W. Dystroglycan binds nerve and muscle agrin. Neuron 1994, 13, 103–115. [Google Scholar] [CrossRef]
- Hopf, C.; Hoch, W. Agrin binding to alpha-dystroglycan. Domains of agrin necessary to induce acetylcholine receptor clustering are overlapping but not identical to the alpha-dystroglycan-binding region. J. Biol. Chem. 1996, 271, 5231–5236. [Google Scholar] [PubMed]
- Gesemann, M.; Cavalli, V.; Denzer, A.J.; Brancaccio, A.; Schumacher, B.; Ruegg, M.A. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron 1996, 16, 755–767. [Google Scholar] [CrossRef]
- Meier, T.; Gesemann, M.; Cavalli, V.; Ruegg, M.A.; Wallace, B.G. AChR phosphorylation and aggregation induced by an agrin fragment that lacks the binding domain for alpha-dystroglycan. EMBO J. 1996, 15, 2625–2631. [Google Scholar] [PubMed]
- Qu, Z.C.; Moritz, E.; Huganir, R.L. Regulation of tyrosine phosphorylation of the nicotinic acetylcholine receptor at the rat neuromuscular junction. Neuron 1990, 4, 367–378. [Google Scholar] [CrossRef]
- Jennings, C.G.; Dyer, S.M.; Burden, S.J. Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases. Proc. Natl. Acad. Sci. USA 1993, 90, 2895–2899. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.K.; Nusse, R. The Frizzled CRD domain is conserved in diverse proteins including several receptor tyrosine kinases. Curr. Biol. 1998, 8, R405–R406. [Google Scholar] [CrossRef]
- Masiakowski, P.; Yancopoulos, G.D. The Wnt receptor CRD domain is also found in MuSK and related orphan receptor tyrosine kinases. Curr. Biol. 1998, 8, R407. [Google Scholar] [CrossRef]
- Valenzuela, D.M.; Stitt, T.N.; DiStefano, P.S.; Rojas, E.; Mattsson, K.; Compton, D.L.; Nunez, L.; Park, J.S.; Stark, J.L.; Gies, D.R.; et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: Expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron 1995, 15, 573–584. [Google Scholar] [CrossRef]
- Burden, S.J.; Yumoto, N.; Zhang, W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a009167. [Google Scholar] [CrossRef] [PubMed]
- Cheusova, T.; Khan, M.A.; Enz, R.; Hashemolhosseini, S. Identification of developmentally regulated expression of MuSK in astrocytes of the rodent retina. J. Neurochem. 2006, 99, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Osta, A.; Tsokas, P.; Pollonini, G.; Landau, E.M.; Blitzer, R.; Alberini, C.M. MuSK expressed in the brain mediates cholinergic responses, synaptic plasticity, and memory formation. J. Neurosci. 2006, 26, 7919–7932. [Google Scholar] [CrossRef] [PubMed]
- Ganju, P.; Walls, E.; Brennan, J.; Reith, A.D. Cloning and developmental expression of Nsk2, a novel receptor tyrosine kinase implicated in skeletal myogenesis. Oncogene 1995, 11, 281–290. [Google Scholar] [PubMed]
- Fu, A.K.; Smith, F.D.; Zhou, H.; Chu, A.H.; Tsim, K.W.; Peng, B.H.; Ip, N.Y. Xenopus muscle-specific kinase: Molecular cloning and prominent expression in neural tissues during early embryonic development. Eur. J. Neurosci. 1999, 11, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Lakshmanan, M.; Swa, H.L.; Chen, J.; Zhang, X.; Ong, Y.S.; Loo, L.S.; Akincilar, S.C.; Gunaratne, J.; Tergaonkar, V.; et al. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat. Commun. 2015, 6, 6184. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512. [Google Scholar] [CrossRef]
- Merlie, J.P.; Sanes, J.R. Concentration of acetylcholine receptor mRNA in synaptic regions of adult muscle fibres. Nature 1985, 317, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Burden, S.J. Synapse-specific gene expression. Trends Genet. 1993, 9, 12–16. [Google Scholar] [CrossRef]
- Glass, D.J.; Bowen, D.C.; Stitt, T.N.; Radziejewski, C.; Bruno, J.; Ryan, T.E.; Gies, D.R.; Shah, S.; Mattsson, K.; Burden, S.J.; et al. Agrin acts via a MuSK receptor complex. Cell 1996, 85, 513–523. [Google Scholar] [CrossRef]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Glass, D.J.; Yancopoulos, G.D.; Sanes, J.R. Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations. J. Cell Biol. 1999, 146, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.; Burden, S.J. The juxtamembrane region of MuSK has a critical role in agrin-mediated signaling. EMBO J. 2000, 19, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Till, J.H.; Becerra, M.; Watty, A.; Lu, Y.; Ma, Y.; Neubert, T.A.; Burden, S.J.; Hubbard, S.R. Crystal structure of the MuSK tyrosine kinase: Insights into receptor autoregulation. Structure 2002, 10, 1187–1196. [Google Scholar] [CrossRef]
- Weston, C.; Gordon, C.; Teressa, G.; Hod, E.; Ren, X.D.; Prives, J. Cooperative regulation by Rac and Rho of agrin-induced acetylcholine receptor clustering in muscle cells. J. Biol. Chem. 2003, 278, 6450–6455. [Google Scholar] [CrossRef] [PubMed]
- Weston, C.; Yee, B.; Hod, E.; Prives, J. Agrin-induced acetylcholine receptor clustering is mediated by the small guanosine triphosphatases Rac and Cdc42. J. Cell Biol. 2000, 150, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Luo, X.; Xie, H.; Peng, H.B. The actin-driven movement and formation of acetylcholine receptor clusters. J. Cell Biol. 2000, 150, 1321–1334. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, R.; Gong, Z.L.; Ma, J.J.; Chan, A.W.; Peng, H.B. The function of cortactin in the clustering of acetylcholine receptors at the vertebrate neuromuscular junction. PLoS ONE 2009, 4, e8478. [Google Scholar] [CrossRef] [PubMed]
- Strochlic, L.; Cartaud, A.; Labas, V.; Hoch, W.; Rossier, J.; Cartaud, J. MAGI-1c: A synaptic MAGUK interacting with muSK at the vertebrate neuromuscular junction. J. Cell Biol. 2001, 153, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Je, H.S.; Young, P.; Gross, J.; Lu, B.; Feng, G. Regulation of synaptic growth and maturation by a synapse-associated E3 ubiquitin ligase at the neuromuscular junction. J. Cell Biol. 2007, 177, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Cartaud, A.; Strochlic, L.; Guerra, M.; Blanchard, B.; Lambergeon, M.; Krejci, E.; Cartaud, J.; Legay, C. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J. Cell Biol. 2004, 165, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.C.; Barzaghi, P.; Ruegg, M.A. Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep. 2004, 5, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hesser, B.A.; Henschel, O.; Witzemann, V. Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol. Cell. Neurosci. 2006, 31, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Shigemoto, K.; Kubo, S.; Maruyama, N.; Hato, N.; Yamada, H.; Jie, C.; Kobayashi, N.; Mominoki, K.; Abe, Y.; Ueda, N.; et al. Induction of myasthenia by immunization against muscle-specific kinase. J. Clin. Investig. 2006, 116, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Xu, K.; Maruta, T.; Oshima, M.; Mosier, D.R.; Atassi, M.Z.; Hoch, W. Myasthenia gravis induced in mice by immunization with the recombinant extracellular domain of rat muscle-specific kinase (MuSK). J. Neuroimmunol. 2006, 175, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Hoch, W.; McConville, J.; Helms, S.; Newsom-Davis, J.; Melms, A.; Vincent, A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med. 2001, 7, 365–368. [Google Scholar] [PubMed]
- Cole, R.N.; Reddel, S.W.; Gervasio, O.L.; Phillips, W.D. Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann. Neurol. 2008, 63, 782–789. [Google Scholar] [PubMed]
- Klooster, R.; Plomp, J.J.; Huijbers, M.G.; Niks, E.H.; Straasheijm, K.R.; Detmers, F.J.; Hermans, P.W.; Sleijpen, K.; Verrips, A.; Losen, M.; et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012, 135, 1081–1101. [Google Scholar] [PubMed]
- Weatherbee, S.D.; Anderson, K.V.; Niswander, L.A. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development 2006, 133, 4993–5000. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Burden, S.J. MuSK controls where motor axons grow and form synapses. Nat. Neurosci. 2008, 11, 19–27. [Google Scholar] [PubMed]
- Zhang, B.; Luo, S.; Wang, Q.; Suzuki, T.; Xiong, W.C.; Mei, L. LRP4 serves as a coreceptor of Agrin. Neuron 2008, 60, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Coldefy, A.S.; Hubbard, S.R.; Burden, S.J. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J. Biol. Chem. 2011, 286, 40624–40630. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Zhang, B.; Gu, S.; Lee, K.; Zhou, J.; Yao, G.; Figueiredo, D.; Perry, K.; Mei, L.; Jin, R. Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 2012, 26, 247–258. [Google Scholar] [PubMed]
- Ohkawara, B.; Cabrera-Serrano, M.; Nakata, T.; Milone, M.; Asai, N.; Ito, K.; Ito, M.; Masuda, A.; Ito, Y.; Engel, A.G.; et al. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising Agrin-mediated MuSK signaling in a position-specific manner. Hum. Mol. Genet. 2014, 23, 1856–1868. [Google Scholar] [PubMed]
- Selcen, D.; Ohkawara, B.; Shen, X.M.; McEvoy, K.; Ohno, K.; Engel, A.G. Impaired Synaptic Development, Maintenance, and Neuromuscular Transmission in LRP4-Related Myasthenia. JAMA Neurol. 2015, 72, 889–896. [Google Scholar] [PubMed]
- Gomez, A.M.; Burden, S.J. The extracellular region of Lrp4 is sufficient to mediate neuromuscular synapse formation. Dev. Dyn. 2011, 240, 2626–2633. [Google Scholar] [PubMed]
- Johnson, E.B.; Hammer, R.E.; Herz, J. Abnormal development of the apical ectodermal ridge and polysyndactyly in Megf7-deficient mice. Hum. Mol. Genet. 2005, 14, 3523–3538. [Google Scholar] [PubMed]
- Duchesne, A.; Gautier, M.; Chadi, S.; Grohs, C.; Floriot, S.; Gallard, Y.; Caste, G.; Ducos, A.; Eggen, A. Identification of a doublet missense substitution in the bovine LRP4 gene as a candidate causal mutation for syndactyly in Holstein cattle. Genomics 2006, 88, 610–621. [Google Scholar] [PubMed]
- Huijbers, M.G.; Zhang, W.; Klooster, R.; Niks, E.H.; Friese, M.B.; Straasheijm, K.R.; Thijssen, P.E.; Vrolijk, H.; Plomp, J.J.; Vogels, P.; et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc. Natl. Acad. Sci. USA 2013, 110, 20783–20788. [Google Scholar] [PubMed]
- Koneczny, I.; Cossins, J.; Waters, P.; Beeson, D.; Vincent, A. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1-3 can disperse preformed agrin-independent AChR clusters. PLoS ONE 2013, 8, e80695. [Google Scholar]
- Li, Y.; Pawlik, B.; Elcioglu, N.; Aglan, M.; Kayserili, H.; Yigit, G.; Percin, F.; Goodman, F.; Nurnberg, G.; Cenani, A.; et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet. 2010, 86, 696–706. [Google Scholar] [PubMed]
- Karner, C.M.; Dietrich, M.F.; Johnson, E.B.; Kappesser, N.; Tennert, C.; Percin, F.; Wollnik, B.; Carroll, T.J.; Herz, J. Lrp4 regulates initiation of ureteric budding and is crucial for kidney formation—A mouse model for Cenani-Lenz syndrome. PLoS ONE 2010, 5, e10418. [Google Scholar]
- Yumoto, N.; Kim, N.; Burden, S.J. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature 2012, 489, 438–442. [Google Scholar] [PubMed]
- Gomez, A.M.; Froemke, R.C.; Burden, S.J. Synaptic plasticity and cognitive function are disrupted in the absence of Lrp4. eLife 2014, 3, e04287. [Google Scholar] [PubMed]
- Pohlkamp, T.; Durakoglugil, M.; Lane-Donovan, C.; Xian, X.; Johnson, E.B.; Hammer, R.E.; Herz, J. Lrp4 domains differentially regulate limb/brain development and synaptic plasticity. PLoS ONE 2015, 10, e0116701. [Google Scholar]
- Sun, X.D.; Li, L.; Liu, F.; Huang, Z.H.; Bean, J.C.; Jiao, H.F.; Barik, A.; Kim, S.M.; Wu, H.; Shen, C.; et al. Lrp4 in astrocytes modulates glutamatergic transmission. Nat. Neurosci. 2016, 19, 1010–1018. [Google Scholar] [PubMed]
- Karakatsani, A.; Marichal, N.; Urban, S.; Kalamakis, G.; Ghanem, A.; Schick, A.; Zhang, Y.; Conzelmann, K.K.; Ruegg, M.A.; Berninger, B.; et al. Neuronal LRP4 regulates synapse formation in the developing CNS. Development 2017, 144, 4604–4615. [Google Scholar] [PubMed]
- Hilgenberg, L.G.; Su, H.; Gu, H.; O’Dowd, D.K.; Smith, M.A. Alpha3Na+/K+-ATPase is a neuronal receptor for agrin. Cell 2006, 125, 359–369. [Google Scholar] [PubMed]
- Wu, H.; Lu, Y.; Shen, C.; Patel, N.; Gan, L.; Xiong, W.C.; Mei, L. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron 2012, 75, 94–107. [Google Scholar] [PubMed]
- Wybenga-Groot, L.E.; Baskin, B.; Ong, S.H.; Tong, J.; Pawson, T.; Sicheri, F. Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell 2001, 106, 745–757. [Google Scholar] [PubMed]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [PubMed]
- Mashima, R.; Hishida, Y.; Tezuka, T.; Yamanashi, Y. The roles of Dok family adapters in immunoreceptor signaling. Immunol. Rev. 2009, 232, 273–285. [Google Scholar] [PubMed]
- Hamuro, J.; Higuchi, O.; Okada, K.; Ueno, M.; Iemura, S.; Natsume, T.; Spearman, H.; Beeson, D.; Yamanashi, Y. Mutations causing DOK7 congenital myasthenia ablate functional motifs in Dok-7. J. Biol. Chem. 2008, 283, 5518–5524. [Google Scholar] [PubMed]
- Bergamin, E.; Hallock, P.T.; Burden, S.J.; Hubbard, S.R. The cytoplasmic adaptor protein Dok7 activates the receptor tyrosine kinase MuSK via dimerization. Mol. Cell 2010, 39, 100–109. [Google Scholar] [PubMed]
- Hallock, P.T.; Xu, C.F.; Park, T.J.; Neubert, T.A.; Curran, T.; Burden, S.J. Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes Dev. 2010, 24, 2451–2461. [Google Scholar] [PubMed]
- Inoue, A.; Setoguchi, K.; Matsubara, Y.; Okada, K.; Sato, N.; Iwakura, Y.; Higuchi, O.; Yamanashi, Y. Dok-7 activates the muscle receptor kinase MuSK and shapes synapse formation. Sci. Signal. 2009, 2, ra7. [Google Scholar] [PubMed]
- Ueta, R.; Tezuka, T.; Izawa, Y.; Miyoshi, S.; Nagatoishi, S.; Tsumoto, K.; Yamanashi, Y. The carboxyl-terminal region of Dok-7 plays a key, but not essential, role in activation of muscle-specific receptor kinase MuSK and neuromuscular synapse formation. J. Biochem. 2017, 161, 269–277. [Google Scholar] [PubMed]
- Muller, J.S.; Herczegfalvi, A.; Vilchez, J.J.; Colomer, J.; Bachinski, L.L.; Mihaylova, V.; Santos, M.; Schara, U.; Deschauer, M.; Shevell, M.; et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain 2007, 130, 1497–1506. [Google Scholar] [PubMed]
- Beeson, D.; Higuchi, O.; Palace, J.; Cossins, J.; Spearman, H.; Maxwell, S.; Newsom-Davis, J.; Burke, G.; Fawcett, P.; Motomura, M.; et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science 2006, 313, 1975–1978. [Google Scholar] [PubMed]
- Engel, A.G. Current status of the congenital myasthenic syndromes. Neuromuscul. Disord. 2012, 22, 99–111. [Google Scholar] [PubMed]
- Yamanashi, Y.; Higuch, O.; Beeson, D. Dok-7/MuSK signaling and a congenital myasthenic syndrome. Acta Myol. 2008, 27, 25–29. [Google Scholar] [PubMed]
- Selcen, D.; Milone, M.; Shen, X.M.; Harper, C.M.; Stans, A.A.; Wieben, E.D.; Engel, A.G. Dok-7 myasthenia: Phenotypic and molecular genetic studies in 16 patients. Ann. Neurol. 2008, 64, 71–87. [Google Scholar] [PubMed]
- Sobel, A.; Heidmann, T.; Hofler, J.; Changeux, J.P. Distinct protein components from Torpedo marmorata membranes carry the acetylcholine receptor site and the binding site for local anesthetics and histrionicotoxin. Proc. Natl. Acad. Sci. USA 1978, 75, 510–514. [Google Scholar] [PubMed]
- Neubig, R.R.; Krodel, E.K.; Boyd, N.D.; Cohen, J.B. Acetylcholine and local anesthetic binding to Torpedo nicotinic postsynaptic membranes after removal of nonreceptor peptides. Proc. Natl. Acad. Sci. USA 1979, 76, 690–694. [Google Scholar] [PubMed]
- Sakmann, B.; Methfessel, C.; Mishina, M.; Takahashi, T.; Takai, T.; Kurasaki, M.; Fukuda, K.; Numa, S. Role of acetylcholine receptor subunits in gating of the channel. Nature 1985, 318, 538–543. [Google Scholar] [PubMed]
- Maimone, M.M.; Merlie, J.P. Interaction of the 43 kd postsynaptic protein with all subunits of the muscle nicotinic acetylcholine receptor. Neuron 1993, 11, 53–66. [Google Scholar] [CrossRef]
- Borges, L.S.; Yechikhov, S.; Lee, Y.I.; Rudell, J.B.; Friese, M.B.; Burden, S.J.; Ferns, M.J. Identification of a motif in the acetylcholine receptor beta subunit whose phosphorylation regulates rapsyn association and postsynaptic receptor localization. J. Neurosci. 2008, 28, 11468–11476. [Google Scholar] [PubMed]
- Ramarao, M.K.; Cohen, J.B. Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn. Proc. Natl. Acad. Sci. USA 1998, 95, 4007–4012. [Google Scholar] [PubMed]
- Ramarao, M.K.; Bianchetta, M.J.; Lanken, J.; Cohen, J.B. Role of rapsyn tetratricopeptide repeat and coiled-coil domains in self-association and nicotinic acetylcholine receptor clustering. J. Biol. Chem. 2001, 276, 7475–7483. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Ramarao, M.K.; Cohen, J.B. Interactions of the rapsyn RING-H2 domain with dystroglycan. J. Biol. Chem. 2001, 276, 24911–24917. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, G.C.; Luo, S.; Yang, Z.; Xiong, W.C.; Mei, L. alpha-Actinin interacts with rapsyn in agrin-stimulated AChR clustering. Mol. Brain 2008, 1, 18. [Google Scholar] [PubMed]
- Li, L.; Cao, Y.; Wu, H.; Ye, X.; Zhu, Z.; Xing, G.; Shen, C.; Barik, A.; Zhang, B.; Xie, X.; et al. Enzymatic Activity of the Scaffold Protein Rapsyn for Synapse Formation. Neuron 2016, 92, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Antolik, C.; Catino, D.H.; O’Neill, A.M.; Resneck, W.G.; Ursitti, J.A.; Bloch, R.J. The actin binding domain of ACF7 binds directly to the tetratricopeptide repeat domains of rapsyn. Neuroscience 2007, 145, 56–65. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, L.D.; Regan, L. TPR proteins: The versatile helix. Trends Biochem. Sci. 2003, 28, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Zeytuni, N.; Zarivach, R. Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 2012, 20, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Gatto, G.J., Jr.; Geisbrecht, B.V.; Gould, S.J.; Berg, J.M. Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5. Nat. Struct. Biol. 2000, 7, 1091–1095. [Google Scholar] [PubMed]
- Gautam, M.; Noakes, P.G.; Mudd, J.; Nichol, M.; Chu, G.C.; Sanes, J.R.; Merlie, J.P. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 1995, 377, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Apel, E.D.; Glass, D.J.; Moscoso, L.M.; Yancopoulos, G.D.; Sanes, J.R. Rapsyn is required for MuSK signaling and recruits synaptic components to a MuSK-containing scaffold. Neuron 1997, 18, 623–635. [Google Scholar] [CrossRef]
- Ohno, K.; Engel, A.G.; Shen, X.M.; Selcen, D.; Brengman, J.; Harper, C.M.; Tsujino, A.; Milone, M. Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome. Am. J. Hum. Genet. 2002, 70, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Marchand, S.; Bignami, F.; Stetzkowski-Marden, F.; Cartaud, J. The myristoylated protein rapsyn is cotargeted with the nicotinic acetylcholine receptor to the postsynaptic membrane via the exocytic pathway. J. Neurosci. 2000, 20, 521–528. [Google Scholar] [PubMed]
- Marchand, S.; Devillers-Thiery, A.; Pons, S.; Changeux, J.P.; Cartaud, J. Rapsyn escorts the nicotinic acetylcholine receptor along the exocytic pathway via association with lipid rafts. J. Neurosci. 2002, 22, 8891–8901. [Google Scholar] [PubMed]
- Moransard, M.; Borges, L.S.; Willmann, R.; Marangi, P.A.; Brenner, H.R.; Ferns, M.J.; Fuhrer, C. Agrin regulates rapsyn interaction with surface acetylcholine receptors, and this underlies cytoskeletal anchoring and clustering. J. Biol. Chem. 2003, 278, 7350–7359. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.B.; Blagden, C.S.; Burden, S.J. Synaptic differentiation is defective in mice lacking acetylcholine receptor beta-subunit tyrosine phosphorylation. Development 2007, 134, 4167–4176. [Google Scholar] [CrossRef] [PubMed]
- Zuber, B.; Unwin, N. Structure and superorganization of acetylcholine receptor-rapsyn complexes. Proc. Natl. Acad. Sci. USA 2013, 110, 10622–10627. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, A.W.; Harris, A.J. Neural influence on acetylcholine receptor clusters in embryonic development of skeletal muscles. Nature 1979, 279, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, W.; Prescott, E.D.; Burden, S.J.; Wang, J.C. DNA topoisomerase IIbeta and neural development. Science 2000, 287, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Arber, S.; William, C.; Li, L.; Tanabe, Y.; Jessell, T.M.; Birchmeier, C.; Burden, S.J. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 2001, 30, 399–410. [Google Scholar] [CrossRef]
- Lin, W.; Burgess, R.W.; Dominguez, B.; Pfaff, S.L.; Sanes, J.R.; Lee, K.F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 2001, 410, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Burden, S.J.; Harris, A.J. Patterning of skeletal muscle. Curr. Opin. Neurobiol. 2002, 12, 100–103. [Google Scholar] [CrossRef]
- Jing, L.; Lefebvre, J.L.; Gordon, L.R.; Granato, M. Wnt signals organize synaptic prepattern and axon guidance through the zebrafish unplugged/MuSK receptor. Neuron 2009, 61, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Messeant, J.; Dobbertin, A.; Girard, E.; Delers, P.; Manuel, M.; Mangione, F.; Schmitt, A.; Le Denmat, D.; Molgo, J.; Zytnicki, D.; et al. MuSK frizzled-like domain is critical for mammalian neuromuscular junction formation and maintenance. J. Neurosci. 2015, 35, 4926–4941. [Google Scholar] [CrossRef] [PubMed]
- Remedio, L.; Gribble, K.D.; Lee, J.K.; Kim, N.; Hallock, P.T.; Delestree, N.; Mentis, G.Z.; Froemke, R.C.; Granato, M.; Burden, S.J. Diverging roles for Lrp4 and Wnt signaling in neuromuscular synapse development during evolution. Genes Dev. 2016, 30, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Tintignac, L.A.; Brenner, H.R.; Ruegg, M.A. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Dominguez, B.; Yang, J.; Aryal, P.; Brandon, E.P.; Gage, F.H.; Lee, K.F. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron 2005, 46, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Kummer, T.T.; Lichtman, J.W.; Sanes, J.R. Agrin promotes synaptic differentiation by counteracting an inhibitory effect of neurotransmitter. Proc. Natl. Acad. Sci. USA 2005, 102, 11088–11093. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.; Ip, F.C.; Fu, W.Y.; Cheung, J.; Wang, J.H.; Yung, W.H.; Ip, N.Y. Aberrant motor axon projection, acetylcholine receptor clustering, and neurotransmission in cyclin-dependent kinase 5 null mice. Proc. Natl. Acad. Sci. USA 2005, 102, 15224–15229. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Qian, L.; Yang, Z.H.; Huang, Y.; Ngo, S.T.; Ruan, N.J.; Wang, J.; Schneider, C.; Noakes, P.G.; Ding, Y.Q.; et al. Rapsyn interaction with calpain stabilizes AChR clusters at the neuromuscular junction. Neuron 2007, 55, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Squire, L.R. Fundamental Neuroscience, 4th ed.; Elsevier; Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2013; Chapter 19. [Google Scholar]
- Kummer, T.T.; Misgeld, T.; Lichtman, J.W.; Sanes, J.R. Nerve-independent formation of a topologically complex postsynaptic apparatus. J. Cell Biol. 2004, 164, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Mittaud, P.; Prescott, E.D.; Fuhrer, C.; Burden, S.J. Src, Fyn, and Yes are not required for neuromuscular synapse formation but are necessary for stabilization of agrin-induced clusters of acetylcholine receptors. J. Neurosci. 2001, 21, 3151–3160. [Google Scholar] [PubMed]
- Grady, R.M.; Zhou, H.; Cunningham, J.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R. Tyrosine-phosphorylated and nonphosphorylated isoforms of alpha-dystrobrevin: Roles in skeletal muscle and its neuromuscular and myotendinous junctions. J. Cell Biol. 2003, 160, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Zhou, H.; Cunningham, J.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R. Maturation and maintenance of the neuromuscular synapse: Genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron 2000, 25, 279–293. [Google Scholar] [CrossRef]
- Escher, P.; Burden, S.J. Synapses form in skeletal muscles lacking neuregulin receptors. Science 2005, 308, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, A.; Burden, S.J. Neuromuscular synapse formation in mice lacking motor neuron- and skeletal muscle-derived Neuregulin-1. J. Neurosci. 2006, 26, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Akaaboune, M.; Gajendran, N.; Martinez-Pena y Valenzuela, I.; Wakefield, S.; Thurnheer, R.; Brenner, H.R. Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of alpha-dystrobrevin. J. Cell Biol. 2011, 195, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Tezuka, T.; Inoue, A.; Hoshi, T.; Weatherbee, S.D.; Burgess, R.W.; Ueta, R.; Yamanashi, Y. The MuSK activator agrin has a separate role essential for postnatal maintenance of neuromuscular synapses. Proc. Natl. Acad. Sci. USA 2014, 111, 16556–16561. [Google Scholar] [CrossRef] [PubMed]
- Gingras, J.; Gawor, M.; Bernadzki, K.M.; Grady, R.M.; Hallock, P.; Glass, D.J.; Sanes, J.R.; Proszynski, T.J. Alpha-Dystrobrevin-1 recruits Grb2 and alpha-catulin to organize neurotransmitter receptors at the neuromuscular junction. J. Cell Sci. 2016, 129, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, M.J.; Burden, S.J. Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep. 2012, 2, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Arimura, S.; Okada, T.; Tezuka, T.; Chiyo, T.; Kasahara, Y.; Yoshimura, T.; Motomura, M.; Yoshida, N.; Beeson, D.; Takeda, S.; et al. Neuromuscular disease. DOK7 gene therapy benefits mouse models of diseases characterized by defects in the neuromuscular junction. Science 2014, 345, 1505–1508. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Ohkawara, B.; Ito, M. Agrin-LRP4-MuSK signaling as a therapeutic target for myasthenia gravis and other neuromuscular disorders. Expert Opin. Ther. Targets 2017, 21, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, S.; Tezuka, T.; Arimura, S.; Tomono, T.; Okada, T.; Yamanashi, Y. DOK7 gene therapy enhances motor activity and life span in ALS model mice. EMBO Mol. Med. 2017, 9, 880–889. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burden, S.J.; Huijbers, M.G.; Remedio, L. Fundamental Molecules and Mechanisms for Forming and Maintaining Neuromuscular Synapses. Int. J. Mol. Sci. 2018, 19, 490. https://doi.org/10.3390/ijms19020490
Burden SJ, Huijbers MG, Remedio L. Fundamental Molecules and Mechanisms for Forming and Maintaining Neuromuscular Synapses. International Journal of Molecular Sciences. 2018; 19(2):490. https://doi.org/10.3390/ijms19020490
Chicago/Turabian StyleBurden, Steven J., Maartje G. Huijbers, and Leonor Remedio. 2018. "Fundamental Molecules and Mechanisms for Forming and Maintaining Neuromuscular Synapses" International Journal of Molecular Sciences 19, no. 2: 490. https://doi.org/10.3390/ijms19020490
APA StyleBurden, S. J., Huijbers, M. G., & Remedio, L. (2018). Fundamental Molecules and Mechanisms for Forming and Maintaining Neuromuscular Synapses. International Journal of Molecular Sciences, 19(2), 490. https://doi.org/10.3390/ijms19020490