Vesicular Axonal Transport is Modified In Vivo by Tau Deletion or Overexpression in Drosophila

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
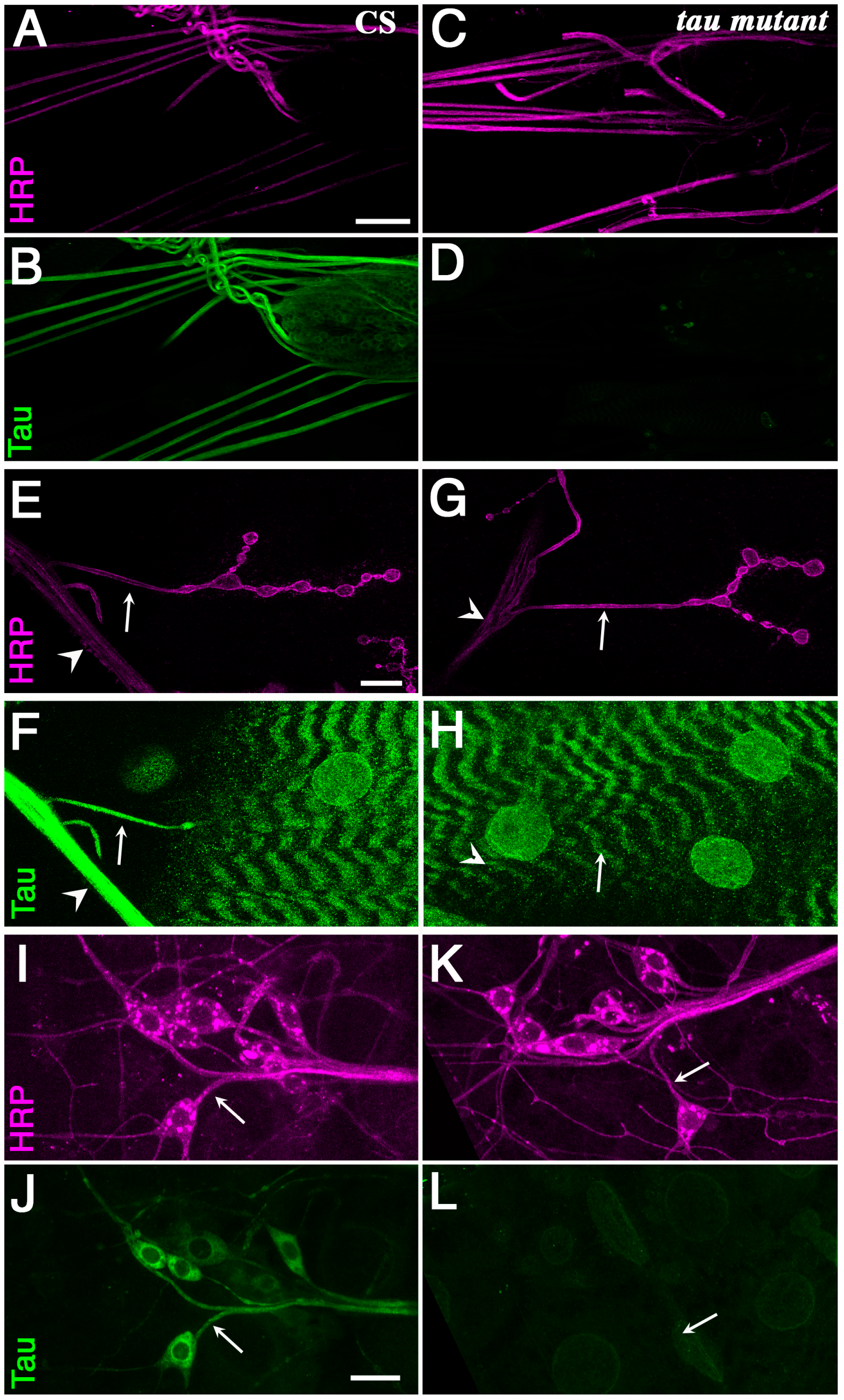
2.1. Tau Is Present in Larval Segmental Nerves
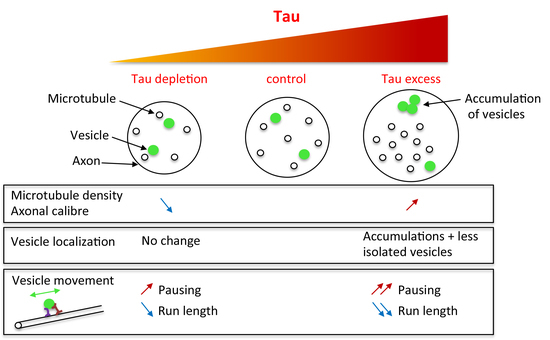
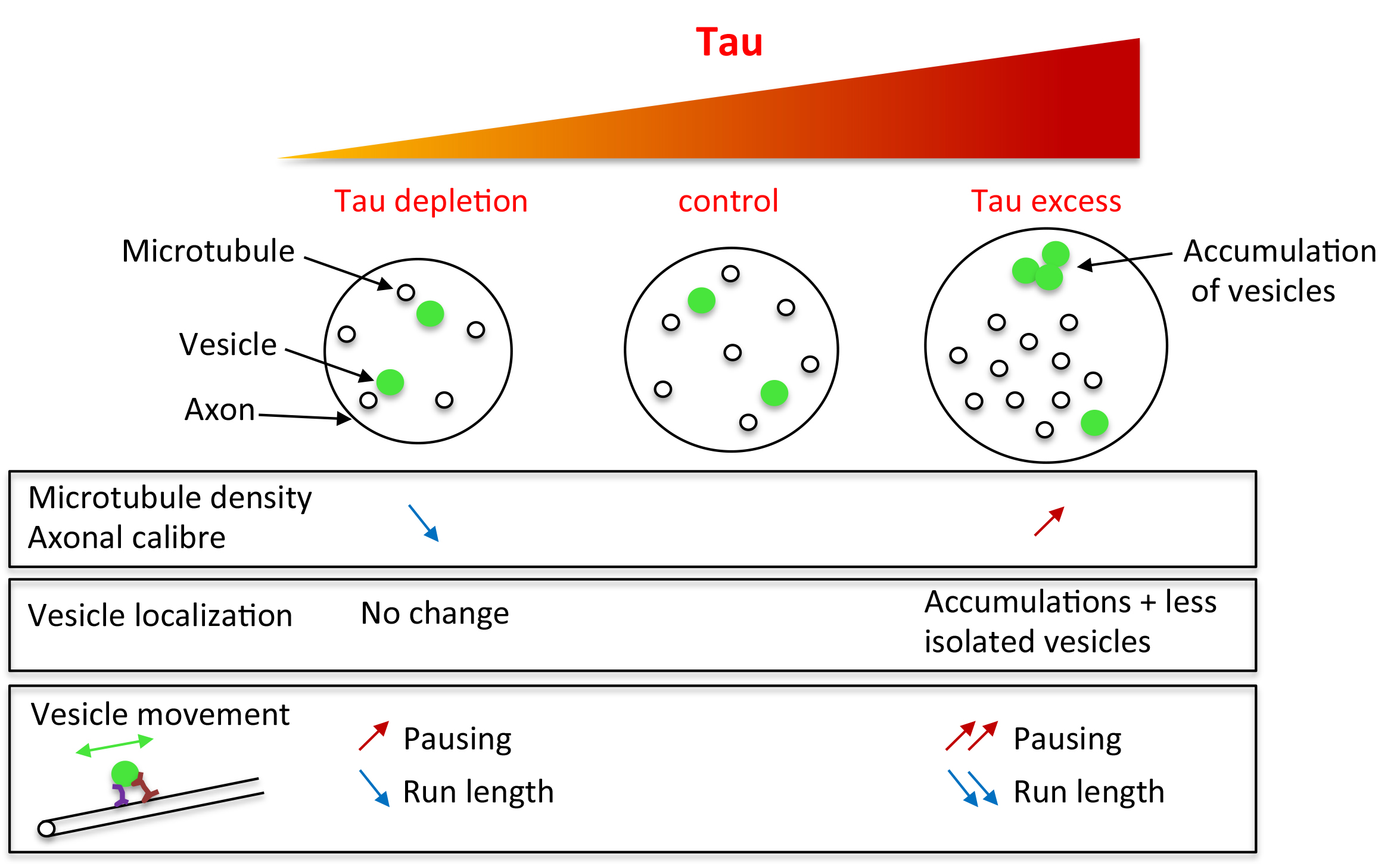
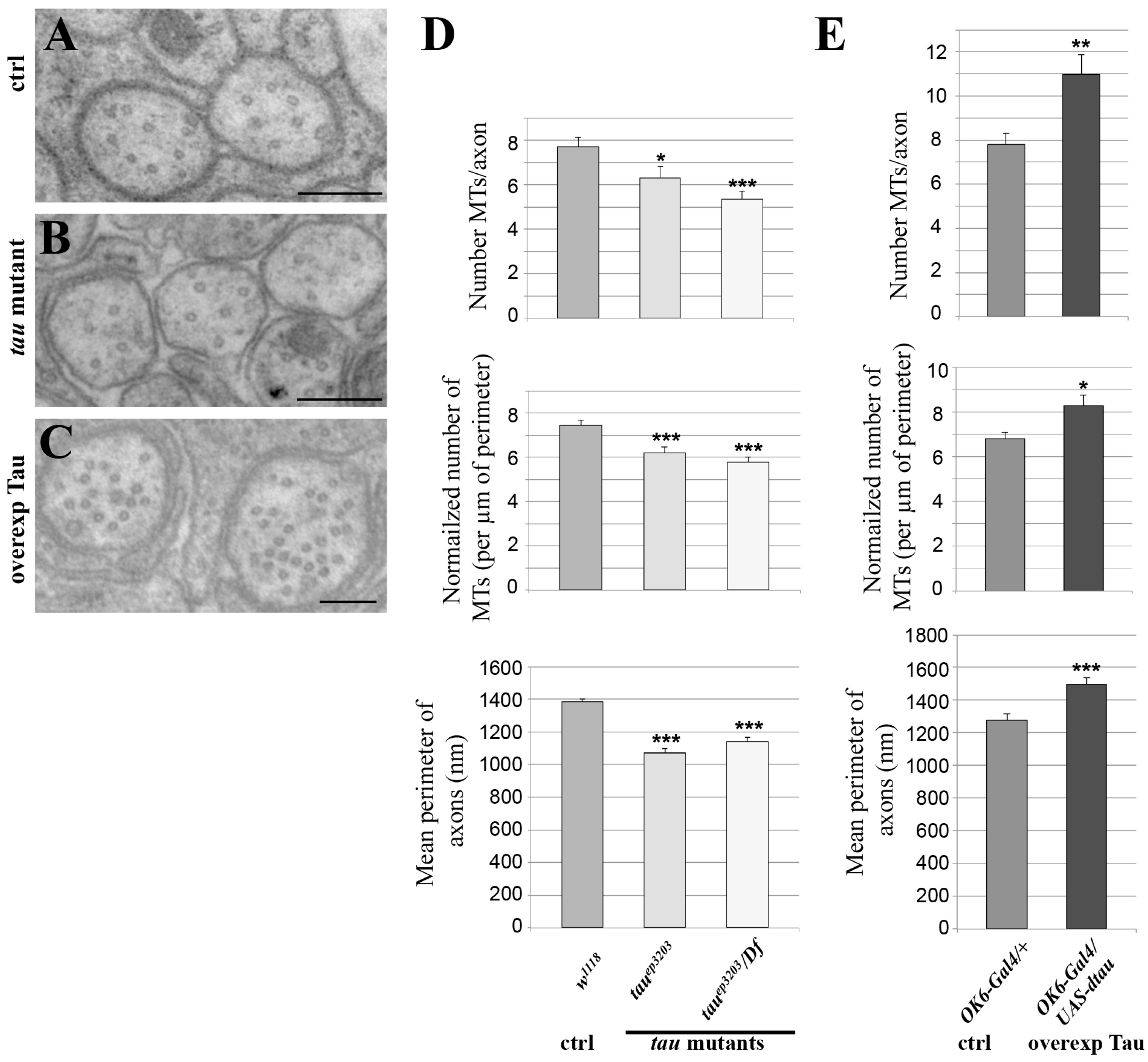
2.2. Tau Controls Microtubule Number in Larval Segmental Axons
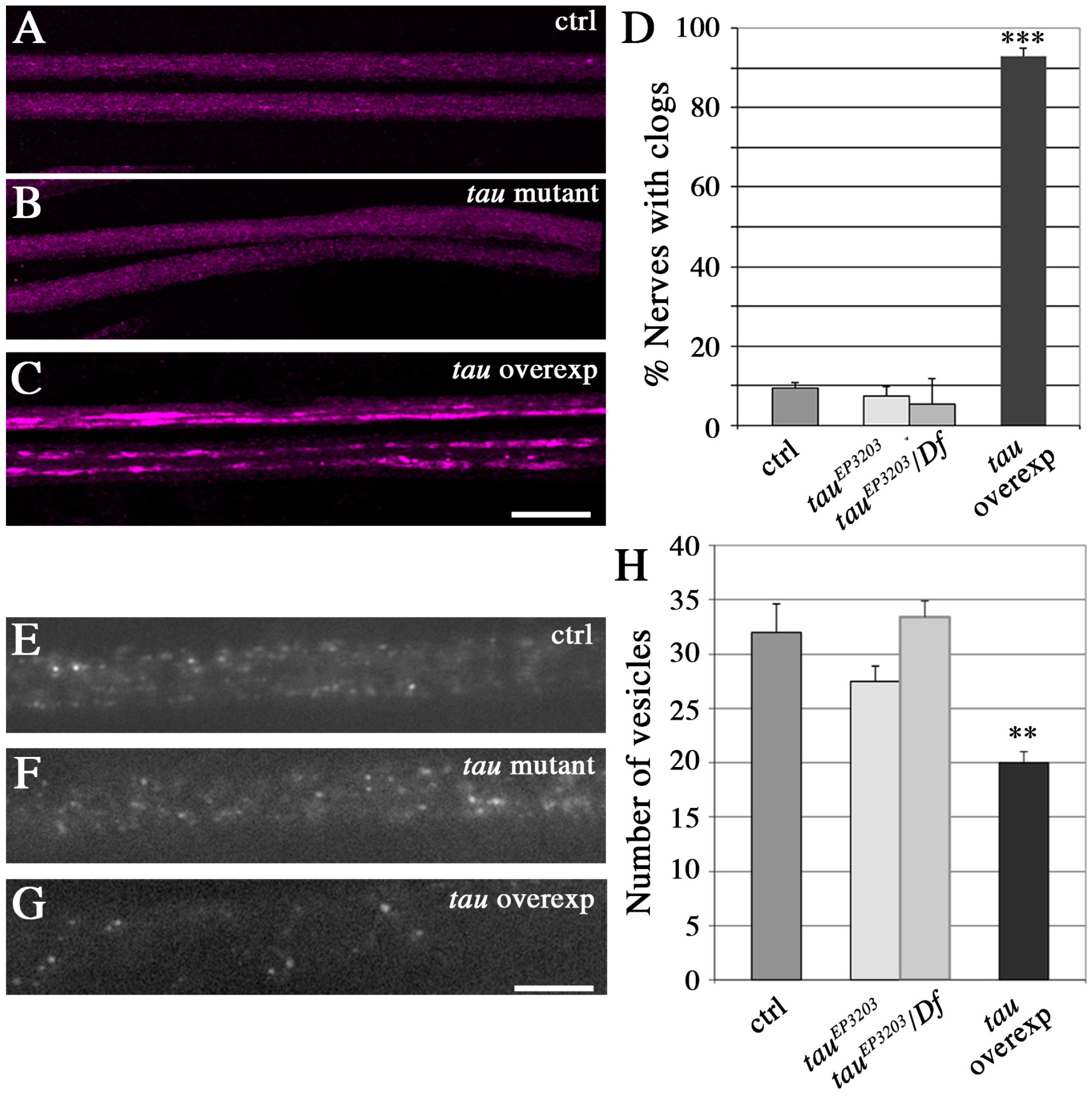
2.3. Tau Depletion Does Not Affect Vesicle Density and Localization in Axons
2.4. Tau Depletion and Tau Excess Differently Affect Vesicle Motion
3. Discussion
4. Materials and Methods
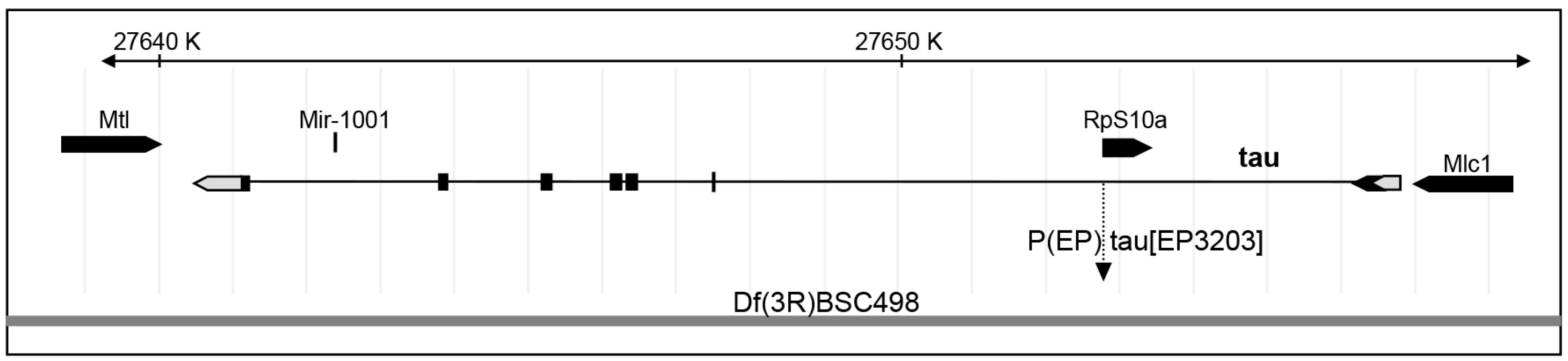
4.1. Fly Stocks
4.2. Immunocytochemistry
4.3. Quantitative PCR Analysis
4.4. Transmission Electron Microscopy
4.5. Non-Invasive Tracking of Vesicles in Segmental Nerves
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HRP | Horse Radish Peroxidase |
| CSP | Cystein String Protein |
| MT | Microtubule |
| MAP | Microtubule-Associated Protein |
References
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005, 6, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Witman, G.B.; Cleveland, D.W.; Weingarten, M.D.; Kirschner, M.W. Tubulin requires tau for growth onto microtubule initiating sites. Proc. Natl. Acad. Sci. USA 1976, 73, 4070–4074. [Google Scholar] [CrossRef] [PubMed]
- Takemura, R.; Okabe, S.; Umeyama, T.; Kanai, Y.; Cowan, N.J.; Hirokawa, N. Increased microtubule stability and alpha tubulin acetylation in cells transfected with microtubule-associated proteins MAP1B, MAP2 or tau. J. Cell Sci. 1992, 103 Pt 4, 953–964. [Google Scholar] [PubMed]
- Maccioni, R.B.; Vera, J.C.; Dominguez, J.; Avila, J. A discrete repeated sequence defines a tubulin binding domain on microtubule-associated protein tau. Arch. Biochem. Biophys. 1989, 275, 568–579. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Kanai, Y.; Chen, J.; Hirokawa, N. Microtubule bundling by tau proteins in vivo: Analysis of functional domains. EMBO J. 1992, 11, 3953–3961. [Google Scholar] [CrossRef]
- Baas, P.W.; Pienkowski, T.P.; Kosik, K.S. Processes induced by tau expression in Sf9 cells have an axon-like microtubule organization. J. Cell Biol. 1991, 115, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kanai, Y.; Cowan, N.J.; Hirokawa, N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 1992, 360, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Gustke, N.; Trinczek, B.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein and interactions with microtubules. Biochemistry 1994, 33, 9511–9522. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 2005, 1739, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Poorkaj, P.; Bird, T.D.; Wijsman, E.; Nemens, E.; Garruto, R.M.; Anderson, L.; Andreadis, A.; Wiederholt, W.C.; Raskind, M.; Schellenberg, G.D. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998, 43, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Nunbhakdi-Craig, V.; Lee, G.; Bloom, G.S.; Mumby, M.C. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron 1996, 17, 1201–1207. [Google Scholar] [CrossRef]
- Alonso, A.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer‘s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Bretteville, A.; Hamdane, M.; Caillet-Boudin, M.L.; Grognet, P.; Bombois, S.; Blum, D.; Delacourte, A.; Pasquier, F.; Vanmechelen, E.; et al. Biochemistry of Tau in Alzheimer's disease and related neurological disorders. Expert Rev. Proteom. 2008, 5, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Jean, D.C.; Baas, P.W. It cuts two ways: Microtubule loss during Alzheimer disease. EMBO J. 2013, 32, 2900–2902. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Luedtke, J.; Kumar, Y.; Biernat, J.; Dawson, H.; Mandelkow, E.; Mandelkow, E.M. Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013, 32, 2920–2937. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Avila, J. Tau aggregates and tau pathology. J. Alzheimers Dis. 2008, 14, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Mandelkow, E. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics 2008, 5, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, S.; Harada, A.; Hirokawa, N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett. 2000, 279, 129–132. [Google Scholar] [CrossRef]
- Yuan, A.; Kumar, A.; Peterhoff, C.; Duff, K.; Nixon, R.A. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci. 2008, 28, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Trinczek, B.; Ebneth, A.; Mandelkow, E.M.; Mandelkow, E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 1999, 112 Pt 14, 2355–2367. [Google Scholar] [PubMed]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer's disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 2003, 24, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Wandosell, F.; Avila, J. Microtubule-associated proteins present in different developmental stages of Drosophila melanogaster. J. Cell. Biochem. 1987, 35, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Heidary, G.; Fortini, M.E. Identification and characterization of the Drosophila tau homolog. Mech. Dev. 2001, 108, 171–178. [Google Scholar] [CrossRef]
- Doerflinger, H.; Benton, R.; Shulman, J.M.; St Johnston, D. The role of PAR-1 in regulating the polarised microtubule cytoskeleton in the Drosophila follicular epithelium. Development 2003, 130, 3965–3975. [Google Scholar] [CrossRef] [PubMed]
- Bolkan, B.J.; Kretzschmar, D. Loss of Tau results in defects in photoreceptor development and progressive neuronal degeneration in Drosophila. Dev. Neurobiol. 2014, 74, 1210–1225. [Google Scholar] [CrossRef] [PubMed]
- Burnouf, S.; Gronke, S.; Augustin, H.; Dols, J.; Gorsky, M.K.; Werner, J.; Kerr, F.; Alic, N.; Martinez, P.; Partridge, L. Deletion of endogenous Tau proteins is not detrimental in Drosophila. Sci. Rep. 2016, 6, 23102. [Google Scholar] [CrossRef] [PubMed]
- Voelzmann, A.; Okenve-Ramos, P.; Qu, Y.; Chojnowska-Monga, M.; Del Cano-Espinel, M.; Prokop, A.; Sanchez-Soriano, N. Tau and spectraplakins promote synapse formation and maintenance through Jun kinase and neuronal trafficking. eLife 2016, 5, e14694. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, S.; Goldstein, L.S. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 2001, 32, 389–401. [Google Scholar] [CrossRef]
- Chee, F.C.; Mudher, A.; Cuttle, M.F.; Newman, T.A.; MacKay, D.; Lovestone, S.; Shepherd, D. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 2005, 20, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Soriano, N.; Tear, G.; Whitington, P.; Prokop, A. Drosophila as a genetic and cellular model for studies on axonal growth. Neural Dev. 2007, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Talmat-Amar, Y.; Parmentier, M.-L.; (Institut de Génomique Fonctionnelle, Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicalem, Montpellier, France). Localization of Tau in the Peripheral Nervous System in Drosophila Larvae. Material not intended for publication. 2012. [Google Scholar]
- Hurd, D.D.; Saxton, W.M. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics 1996, 144, 1075–1085. [Google Scholar] [PubMed]
- Gindhart, J.G., Jr.; Desai, C.J.; Beushausen, S.; Zinn, K.; Goldstein, L.S. Kinesin light chains are essential for axonal transport in Drosophila. J. Cell Biol. 1998, 141, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Shepherd, D.; Newman, T.A.; Mildren, P.; Jukes, J.P.; Squire, A.; Mears, A.; Drummond, J.A.; Berg, S.; MacKay, D.; et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 2004, 9, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ubhi, K.K.; Shaibah, H.; Newman, T.A.; Shepherd, D.; Mudher, A. A comparison of the neuronal dysfunction caused by Drosophila tau and human tau in a Drosophila model of tauopathies. Invert. Neurosci. 2007, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, D.; Collins, C.A.; Bhat, P.; Barkus, R.V.; Diantonio, A.; Saxton, W.M. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr. Biol. 2007, 17, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Talmat-Amar, Y.; Arribat, Y.; Redt-Clouet, C.; Feuillette, S.; Bouge, A.L.; Lecourtois, M.; Parmentier, M.L. Important neuronal toxicity of microtubule-bound Tau in vivo in Drosophila. Hum. Mol. Genet. 2011, 20, 3738–3745. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulos, I.; Galas, M.C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.M.; Mandelkow, E.; et al. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 91. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Teng, J.; Harada, A.; Hirokawa, N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 2000, 150, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, J.; Takei, Y.; Harada, A.; Nakata, T.; Chen, J.; Hirokawa, N. Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J. Cell Biol. 2001, 155, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belanger, D.; Farah, C.A.; Nguyen, M.D.; Lauzon, M.; Cornibert, S.; Leclerc, N. The projection domain of MAP2b regulates microtubule protrusion and process formation in Sf9 cells. J. Cell Sci. 2002, 115 Pt 7, 1523–1539. [Google Scholar] [PubMed]
- Goldstein, L.S.; Gunawardena, S. Flying through the drosophila cytoskeletal genome. J. Cell Biol. 2000, 150, F63–F68. [Google Scholar] [CrossRef] [PubMed]
- Stephan, R.; Goellner, B.; Moreno, E.; Frank, C.A.; Hugenschmidt, T.; Genoud, C.; Aberle, H.; Pielage, J. Hierarchical microtubule organization controls axon caliber and transport and determines synaptic structure and stability. Dev. Cell 2015, 33, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of tau protein with the dynactin complex. EMBO J. 2007, 26, 4546–4554. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.R.; Berger, F.; Berger, C.L.; Hendricks, A.G. Tau directs intracellular trafficking by regulating the forces exerted by kinesin and dynein teams. Traffic 2018, 19, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Erickson, R.P.; Jia, Z.; Gross, S.P.; Yu, C.C. How molecular motors are arranged on a cargo is important for vesicular transport. PLoS Comput. Biol. 2011, 7, e1002032. [Google Scholar] [CrossRef] [PubMed]
- Vershinin, M.; Carter, B.C.; Razafsky, D.S.; King, S.J.; Gross, S.P. Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. USA 2007, 104, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Usardi, A.; Evans, C.J.; Philpott, K.L.; Noble, W.; Hanger, D.P. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol. Aging 2012, 33, 431.e27–431.e38. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef] [PubMed]
- Seitz, A.; Kojima, H.; Oiwa, K.; Mandelkow, E.M.; Song, Y.H.; Mandelkow, E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002, 21, 4896–4905. [Google Scholar] [CrossRef] [PubMed]
- Tien, N.W.; Wu, G.H.; Hsu, C.C.; Chang, C.Y.; Wagner, O.I. Tau/PTL-1 associates with kinesin-3 KIF1A/UNC-104 and affects the motor‘s motility characteristics in C. elegans neurons. Neurobiol. Dis. 2011, 43, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Pigino, G.; Mizuno, N.; Kikkawa, M.; Brady, S.T. Tau binding to microtubules does not directly affect microtubule-based vesicle motility. J. Neurosci. Res. 2007, 85, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Huang, J.; Cao, D.; Yang, G.; Liu, W.; Lu, H.; Guo, A. Study of tauopathies by comparing Drosophila and human tau in Drosophila. Cell Tissue Res. 2007, 329, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Mershin, A.; Pavlopoulos, E.; Fitch, O.; Braden, B.C.; Nanopoulos, D.V.; Skoulakis, E.M. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn. Mem. 2004, 11, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Gistelinck, M.; Lambert, J.C.; Callaerts, P.; Dermaut, B.; Dourlen, P. Drosophila models of tauopathies: What have we learned? Int. J. Alzheimers Dis. 2012, 2012, 970980. [Google Scholar] [PubMed]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Aberle, H.; Haghighi, A.P.; Fetter, R.D.; McCabe, B.D.; Magalhaes, T.R.; Goodman, C.S. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron 2002, 33, 545–558. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talmat-Amar, Y.; Arribat, Y.; Parmentier, M.-L. Vesicular Axonal Transport is Modified In Vivo by Tau Deletion or Overexpression in Drosophila. Int. J. Mol. Sci. 2018, 19, 744. https://doi.org/10.3390/ijms19030744
Talmat-Amar Y, Arribat Y, Parmentier M-L. Vesicular Axonal Transport is Modified In Vivo by Tau Deletion or Overexpression in Drosophila. International Journal of Molecular Sciences. 2018; 19(3):744. https://doi.org/10.3390/ijms19030744
Chicago/Turabian StyleTalmat-Amar, Yasmina, Yoan Arribat, and Marie-Laure Parmentier. 2018. "Vesicular Axonal Transport is Modified In Vivo by Tau Deletion or Overexpression in Drosophila" International Journal of Molecular Sciences 19, no. 3: 744. https://doi.org/10.3390/ijms19030744
APA StyleTalmat-Amar, Y., Arribat, Y., & Parmentier, M. -L. (2018). Vesicular Axonal Transport is Modified In Vivo by Tau Deletion or Overexpression in Drosophila. International Journal of Molecular Sciences, 19(3), 744. https://doi.org/10.3390/ijms19030744