The Role of mTOR in Neuroendocrine Tumors: Future Cornerstone of a Winning Strategy?

 , ,
, , 
Abstract
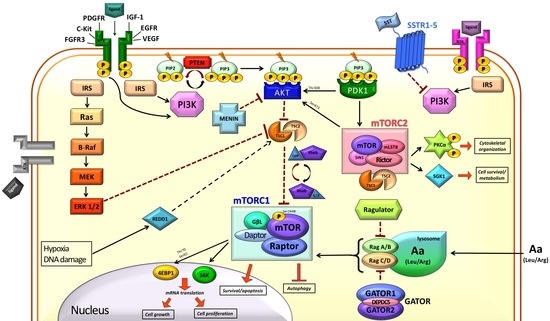
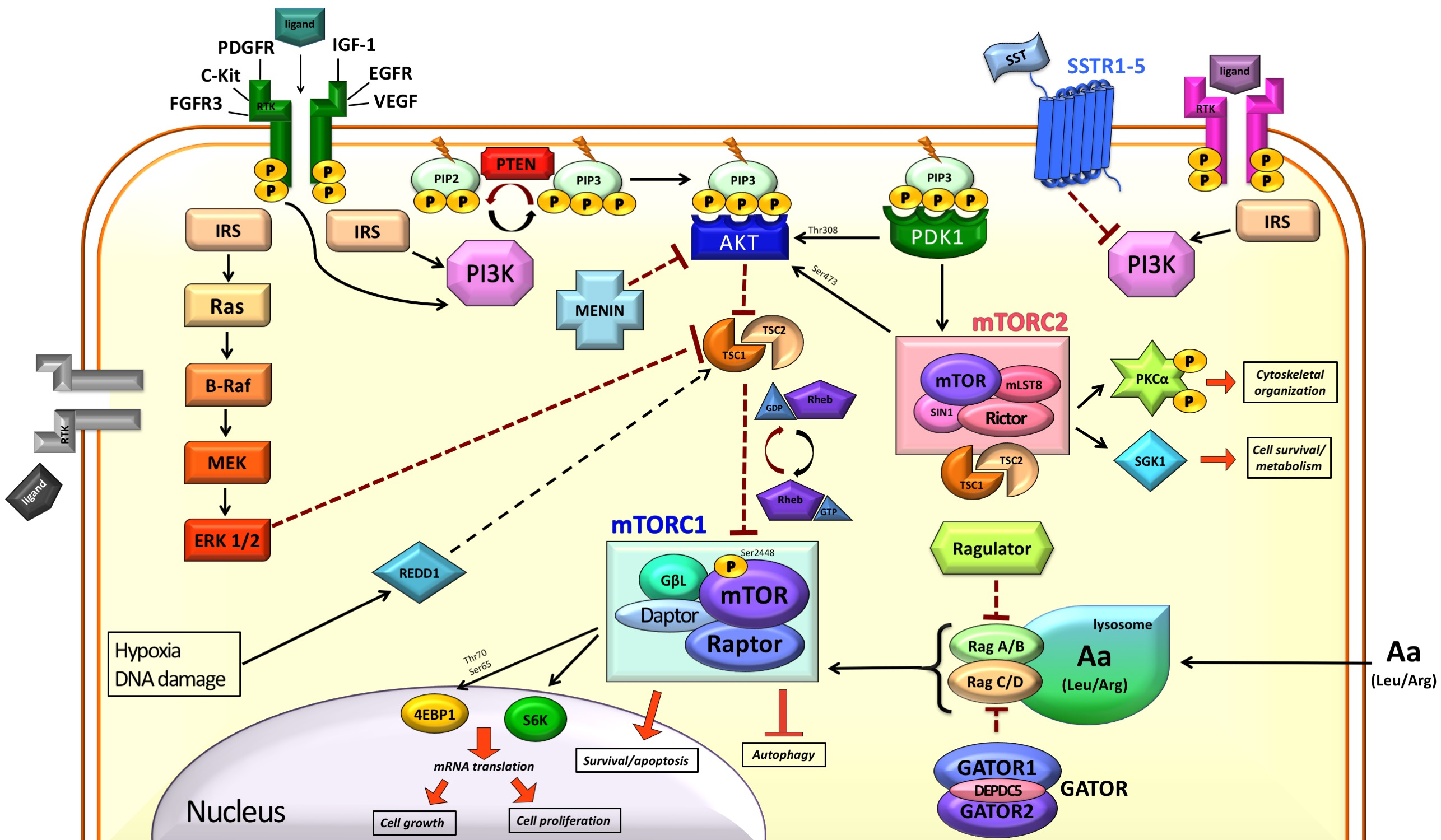
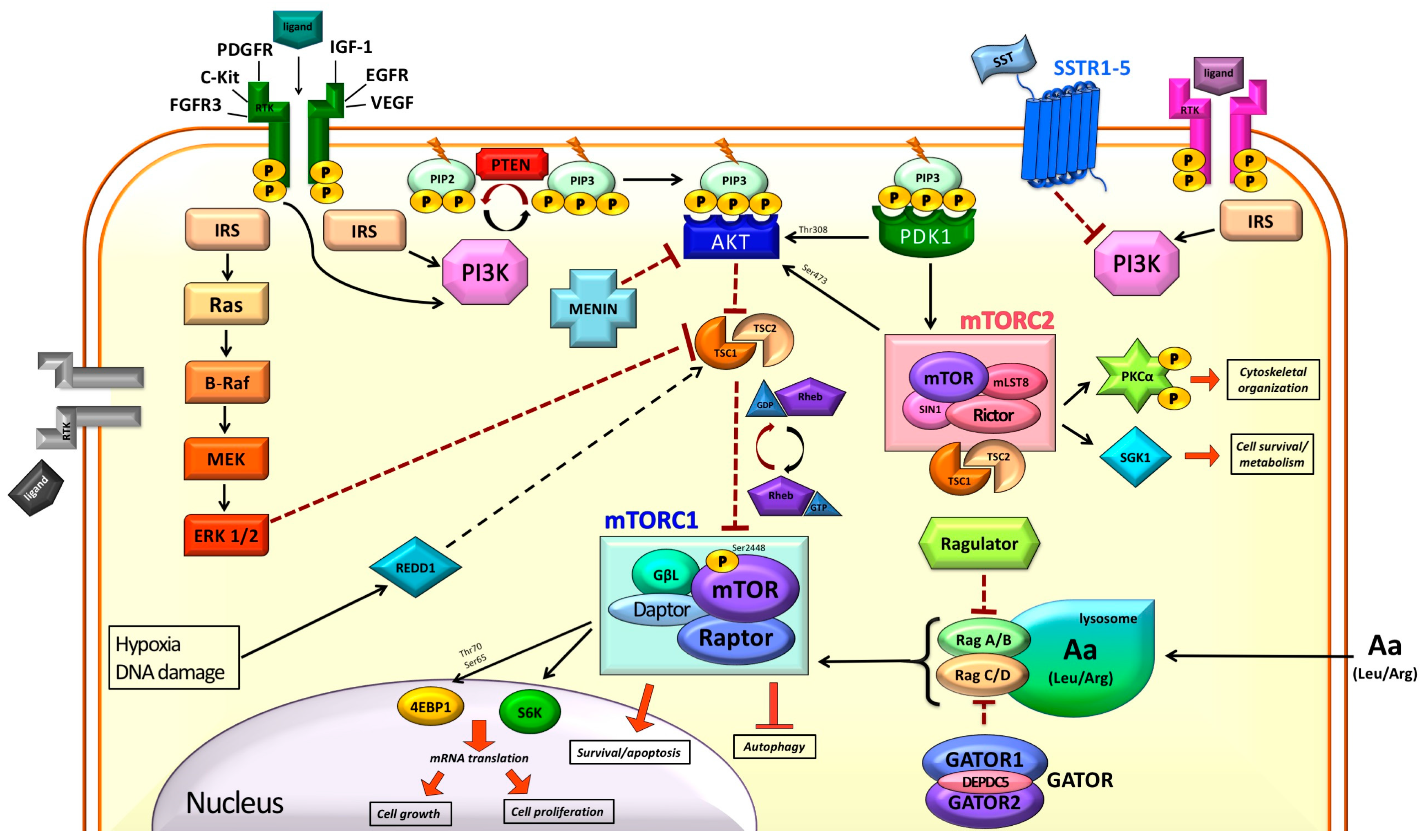
:
1. Introduction
2. mTOR in the Normal Cell
2.1. Modulation of mTOR Activity
2.2. Downstream of mTORC1
3. mTOR and Cancer
4. mTOR and NETs
4.1. Genetic Alterations
4.2. Expression Studies
4.3. Effects of mTOR Inhibition
5. Clinical Trials
6. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 4EBP1 | Eukaryotic translation initiation factor 4E binding protein 1 |
| Akt | Protein kinase B |
| AMPK | Adenosine monophosphate-activated protein kinase |
| c-KIT | KIT Proto-Oncogene Receptor Tyrosine Kinase |
| DEPDC5 | DEP domain containing 5 |
| EGFR | Epidermal growth factor receptor |
| eIF4E | Eukaryotic translation initiation factor 4E |
| ERK1/2 | Extracellular-signal-regulated kinase 1/2 |
| EWSR1 | Ewing Sarcoma Breakpoint Region 1 |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor 3 |
| GAP | GTPase activating protein |
| GATOR | GAP activity towards Rag |
| GTP | Guanosine triphosphate |
| HIF1a | Hypoxia inducible factor 1alfa |
| IGF-1 | Insulin-like growth factor-1 |
| IGF-1R | Insulin-like growth factor-1 receptor |
| IHC | Immunohistochemistry |
| MAPK | Mitogen-activated protein kinase |
| mTOR | Mechanistic target of rapamycin |
| NET | Neuroendocrine tumor |
| NET | Neuroendocrine tumor |
| ORR | Objective response rate |
| OS | Overall Survival |
| PDGFR | Platelet-derived growth factor receptor |
| PDK1 | 3-phosphoinositide-dependent kinase-1 |
| PFS | Progression-free survival |
| PI3K | Phosphoinositide-3-kinase |
| PIP2 | Phospatidylinositol-4,5,-bisphospate |
| PIP3 | Phospatidylinositol-3,4,5,-trisphospate |
| PKB | Protein kinase B |
| PKCa | Protein kinase C-alfa |
| PPAR-y | Peroxisome proliferator-activated recetpor y |
| PSPN | Persephin |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 10 |
| Rag | Ras-related GTPase |
| Raptor | Regulatory-associated protein of mammalian target of rapamycin |
| RET | Rearranged during transfection |
| Rheb | Ras homolog enriched in brain |
| Rictor | Rapamycin-insensitive companion of mammalian target of rapamycin |
| RSK1 | Ribosomal S6 kinase |
| S6K1 | S6 kinase 1 |
| SGK1 | Serum- and glucocorticoid-induced protein kinase 1 |
| SREBP1/2 | Sterol regulatory element-binding protein 1/2 |
| SSA | Somatostatin analog |
| SSTR1-5 | Somatostatin receptor 1-5 |
| SSTR1-5 | Somatostatin receptor 1-5 |
| TNFa | Tumor necrosis factor alfa |
| TSC | Tuberous sclerosis |
| TTP | Time to tumor progression |
| VEGF | Vascular endothelial growth factor |
| Wnt | Wingless-related integration site |
References
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.-N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef] [PubMed]
- Hallet, J.; Law, C.H.L.; Cukier, M.; Saskin, R.; Liu, N.; Singh, S. Exploring the rising incidence of neuroendocrine tumors: A population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer 2015, 121, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Lawrence, M.S.; Getz, G.; Bader, G.D.; Ding, L.; et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013, 3, 2650. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Dalai, I.; Barbi, S.; Beghelli, S.; Falconi, M.; della Peruta, M.; Piemonti, L.; Capurso, G.; Di Florio, A.; delle Fave, G.; et al. Pancreatic endocrine tumors: Expression profiling evidences a role for AKT-mTOR pathway. J. Clin. Oncol. 2010, 28, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science (80-) 2011, 331. [Google Scholar] [CrossRef] [PubMed]
- Kasajima, A.; Pavel, M.; Darb-Esfahani, S.; Noske, A.; Stenzinger, A.; Sasano, H.; Dietel, M.; Denkert, C.; Röcken, C.; Wiedenmann, B.; et al. mTOR expression and activity patterns in gastroenteropancreatic neuroendocrine tumours. Endocr. Relat. Cancer 2011, 18, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Dave, B. The pivotal role of mammalian target of rapamycin inhibition in the treatment of patients with neuroendocrine tumors. Cancer Med. 2016, 5, 2953–2964. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Svejda, B.; Kidd, M.; Kazberouk, A.; Lawrence, B.; Pfragner, R.; Modlin, I.M. Limitations in small intestinal neuroendocrine tumor therapy by mTor kinase inhibition reflect growth factor-mediated PI3K feedback loop activation via ERK1/2 and AKT. Cancer 2011, 117, 4141–4154. [Google Scholar] [CrossRef] [PubMed]
- Cingarlini, S.; Bonomi, M.; Corbo, V.; Scarpa, A.; Tortora, G. Profiling mTOR pathway in neuroendocrine tumors. Target. Oncol. 2012, 7, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.W.; Nozawa, H.; Hanahan, D. Survival Benefit with Proapoptotic Molecular and Pathologic Responses from Dual Targeting of Mammalian Target of Rapamycin and Epidermal Growth Factor Receptor in a Preclinical Model of Pancreatic Neuroendocrine Carcinogenesis. J. Clin. Oncol. 2010, 28, 4425–4433. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-F.; Kuo, H.-P.; Chen, C.-T.; Hsu, J.-M.; Chou, C.-K.; Wei, Y.; Sun, H.-L.; Li, L.-Y.; Ping, B.; Huang, W.-C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Haar, E.V.; Lee, S.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Bjornsti, M.-A.; Houghton, P.J. The tor pathway: A target for cancer therapy. Nat. Rev. Cancer 2004, 4, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the mTOR Signaling Network for Cancer Therapy. J. Clin. Oncol. 2009, 27, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Land, S.C.; Tee, A.R. Hypoxia-inducible Factor 1α Is Regulated by the Mammalian Target of Rapamycin (mTOR) via an mTOR Signaling Motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubin-de-Celis, S.; Abdallah, Z.; Kinch, L.; Grishin, N.V.; Brugarolas, J.; Zhang, X. Structural analysis and functional implications of the negative mTORC1 regulator REDD1. Biochemistry 2010, 49, 2491–2501. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, E. Rag GTPase in amino acid signaling. Amino Acids 2016, 48, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Blenis, J. Target of rapamycin (TOR): An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004, 23, 3151–3171. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Righi, L.; Volante, M.; Rapa, I.; Tavaglione, V.; Inzani, F.; Pelosi, G.; Papotti, M. Mammalian target of rapamycin signaling activation patterns in neuroendocrine tumors of the lung. Endocr. Relat. Cancer 2010, 17, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.; Parra, J.-L.; Beugnet, A.; Proud, C.G. The C terminus of initiation factor 4E-binding protein 1 contains multiple regulatory features that influence its function and phosphorylation. Mol. Cell. Biol. 2003, 23, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Heesom, K.J.; Gampel, A.; Mellor, H.; Denton, R.M. Cell cycle-dependent phosphorylation of the translational repressor eIF-4E binding protein-1 (4E-BP1). Curr. Biol. 2001, 11, 1374–1379. [Google Scholar] [CrossRef]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ogawa, W.; Emi, A.; Hayashi, K.; Senga, Y.; Nomura, K.; Hara, K.; Yu, D.; Kasuga, M. Role of S6K1 in regulation of SREBP1c expression in the liver. Biochem. Biophys. Res. Commun. 2011, 412, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Chen, J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Huang, J.; Düvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.-L.; Manning, B.D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE 2009, 4, e6189. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 2010, 17, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 2004, 10, 331S–336S. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Pieterman, C.R.C.; Conemans, E.B.; Dreijerink, K.M.A.; de Laat, J.M.; Timmers, H.T.M.; Vriens, M.R.; Valk, G.D. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: Natural history and function of menin in tumorigenesis. Endocr. Relat. Cancer 2014, 21, R121–R142. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Missiaglia, E.; Antonello, D.; Zamó, A.; Zamboni, G.; Corleto, V.; Falconi, M.; Scarpa, A. Role of disease-causing genes in sporadic pancreatic endocrine tumors: MEN1 and VHL. Genes Chromosom. Cancer 2001, 32, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Perren, A.; Wiesli, P.; Schmid, S.; Montani, M.; Schmitt, A.; Schmid, C.; Moch, H.; Komminoth, P. Pancreatic endocrine tumors are a rare manifestation of the neurofibromatosis type 1 phenotype: Molecular analysis of a malignant insulinoma in a NF-1 patient. Am. J. Surg. Pathol. 2006, 30, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Kawabata, Y.; Hari, Y.; Imaoka, H.; Ishikawa, N.; Yano, S.; Maruyama, R.; Tajima, Y. A case of pancreatic neuroendocrine tumor in a patient with neurofibromatosis-1. World J. Surg. Oncol. 2012, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.P.; Korf, B.R.; Theos, A. Neurofibromatosis type 1. J. Am. Acad. Dermatol. 2009, 61. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, S.; van Diemen-Steenvoorde, R.; Akkersdijk, W.L.; Bax, N.M.; Ariyurek, Y.; Hermans, C.J.; van Nieuwenhuizen, O.; Nikkels, P.G.; Lindhout, D.; Halley, D.J.; et al. Malignant pancreatic tumour within the spectrum of tuberous sclerosis complex in childhood. Eur. J. Pediatr. 1999, 158, 284–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, A.; Hedgire, S.; Deshpande, V.; Stemmer-Rachamimov, A.; Harisinghani, M.; Ferrone, C.; Shah, U.; Thiele, E. Pancreatic neuroendocrine tumors in patients with tuberous sclerosis complex. Clin. Genet. 2012, 82, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Francalanci, P.; Diomedi-Camassei, F.; Purificato, C.; Santorelli, F.M.; Giannotti, A.; Dominici, C.; Inserra, A.; Boldrini, R. Malignant pancreatic endocrine tumor in a child with tuberous sclerosis. Am. J. Surg. Pathol. 2003, 27, 1386–1389. [Google Scholar] [CrossRef] [PubMed]
- Blansfield, J.A.; Choyke, L.; Morita, S.Y.; Choyke, P.L.; Pingpank, J.F.; Alexander, H.R.; Seidel, G.; Shutack, Y.; Yuldasheva, N.; Eugeni, M.; et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 2007, 142. [Google Scholar] [CrossRef] [PubMed]
- Langer, S.W.; Ringholm, L.; Dali, C.I.; Petersen, R.H.; Rasmussen, Å.K.; Gerdes, A.-M.; Federspiel, B.; Knigge, U.P. Cowden Syndrome and Concomitant Pulmonary Neuroendocrine Tumor: A Presentation of Two Cases. Case Rep. Med. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Neychev, V.; Sadowski, S.M.; Zhu, J.; Allgaeuer, M.; Kilian, K.; Meltzer, P.; Kebebew, E. Neuroendocrine Tumor of the Pancreas as a Manifestation of Cowden Syndrome: A Case Report. J. Clin. Endocrinol. Metab. 2016, 101, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ignat, A.; Axiotis, C.A. Differential expression of the PTEN tumor suppressor protein in fetal and adult neuroendocrine tissues and tumors: Progressive loss of PTEN expression in poorly differentiated neuroendocrine neoplasms. Appl. Immunohistochem. Mol. Morphol. AIMM 2002, 10, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Toumpanakis, C.G.; Caplin, M.E. Molecular genetics of gastroenteropancreatic neuroendocrine tumors. Am. J. Gastroenterol. 2008, 103, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, M.; Poteryaev, D.; Yu, L.; Arumäe, U.; Timmusk, T.; Bongarzone, I.; Aiello, A.; Pierotti, M.A.; Airaksinen, M.S.; Saarma, M. Human Glial Cell Line-derived Neurotrophic Factor Receptor α4 Is the Receptor for Persephin and Is Predominantly Expressed in Normal and Malignant Thyroid Medullary Cells. J. Biol. Chem. 2001, 276, 9344–9351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, M. PI3K/AKT/mTOR pathway in pulmonary carcinoid tumours. Oncol. Lett. 2017, 14, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ozawa, A.; Zaman, S.; Prasad, N.B.; Chandrasekharappa, S.C.; Agarwal, S.K.; Marx, S.J. The tumor suppressor protein menin inhibits AKT activation by regulating its cellular localization. Cancer Res. 2011, 71, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Miki, Y.; Ono, K.; Akahira, J.; Suzuki, T.; Ishida, K.; Watanabe, M.; Sasano, H. Novel classification based on immunohistochemistry combined with hierarchical clustering analysis in non-functioning neuroendocrine tumor patients. Cancer Sci. 2010, 101, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Adhikari, L.J.; Lloyd, R.V.; Rubin, J.; Haluska, P.; Carboni, J.M.; Gottardis, M.M.; Ames, M.M. Molecular markers for novel therapies in neuroendocrine (carcinoid) tumors. Endocr. Relat. Cancer 2010, 17, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Komori, Y.; Yada, K.; Ohta, M.; Uchida, H.; Iwashita, Y.; Fukuzawa, K.; Kashima, K.; Yokoyama, S.; Inomata, M.; Kitano, S. Mammalian target of rapamycin signaling activation patterns in pancreatic neuroendocrine tumors. J. Hepato-Biliary-Pancreat. Sci. 2014, 21, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G.; Ceccarelli, C.; Brighi, N.; Maggio, I.; Santini, D.; Mosconi, C.; Ricci, C.; Biasco, G.; Campana, D. Determination of Mammalian Target of Rapamycin Hyperactivation as Prognostic Factor in Well-Differentiated Neuroendocrine Tumors. Gastroenterol. Res. Pract. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shida, T.; Kishimoto, T.; Furuya, M.; Nikaido, T.; Koda, K.; Takano, S.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Ohtsuka, M.; et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother. Pharmacol. 2010, 65, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Duran, I.; Kortmansky, J.; Singh, D.; Hirte, H.; Kocha, W.; Goss, G.; Le, L.; Oza, A.; Nicklee, T.; Ho, J.; et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br. J. Cancer 2006, 95, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Perren, A.; Komminoth, P.; Saremaslani, P.; Matter, C.; Feurer, S.; Lees, J.A.; Heitz, P.U.; Eng, C. Mutation and Expression Analyses Reveal Differential Subcellular Compartmentalization of PTEN in Endocrine Pancreatic Tumors Compared to Normal Islet Cells. Am. J. Pathol. 2000, 157, 1097–1103. [Google Scholar] [CrossRef]
- Grozinsky-Glasberg, S.; Franchi, G.; Teng, M.; Leontiou, C.A.; Ribeiro de Oliveira, A.; Dalino, P.; Salahuddin, N.; Korbonits, M.; Grossman, A.B. Octreotide and the mTOR inhibitor RAD001 (everolimus) block proliferation and interact with the Akt-mTOR-p70S6K pathway in a neuro-endocrine tumour cell Line. Neuroendocrinology 2008, 87, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, K.; von Rüden, J.; Brand, S.; Göke, B.; Lichtl, J.; Spöttl, G.; Auernhammer, C.J. Compensatory activation of Akt in response to mTOR and Raf inhibitors—A rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010, 295, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Zatelli, M.C.; Minoia, M.; Martini, C.; Tagliati, F.; Ambrosio, M.R.; Schiavon, M.; Buratto, M.; Calabrese, F.; Gentilin, E.; Cavallesco, G.; et al. Everolimus as a new potential antiproliferative agent in aggressive human bronchial carcinoids. Endocr. Relat. Cancer 2010, 17, 719–729. [Google Scholar] [CrossRef]
- Yao, J.C.; Lombard-Bohas, C.; Baudin, E.; Kvols, L.K.; Rougier, P.; Ruszniewski, P.; Hoosen, S.; St Peter, J.; Haas, T.; Lebwohl, D.; et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J. Clin. Oncol. 2010, 28, 69–76. [Google Scholar] [CrossRef]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Hörsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet (Lond. Engl.) 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Pavel, M.E.; Baudin, E.; Öberg, K.E.; Hainsworth, J.D.; Voi, M.; Rouyrre, N.; Peeters, M.; Gross, D.J.; Yao, J.C. Efficacy of everolimus plus octreotide LAR in patients with advanced neuroendocrine tumor and carcinoid syndrome: Final overall survival from the randomized, placebo-controlled phase 3 RADIANT-2 study. Ann. Oncol. 2017, 28, 1569–1575. [Google Scholar] [CrossRef]
- Ferolla, P.; Brizzi, M.P.; Meyer, T.; Mansoor, W.; Mazieres, J.; Do Cao, C.; Léna, H.; Berruti, A.; Damiano, V.; Buikhuisen, W.; et al. Efficacy and safety of long-acting pasireotide or everolimus alone or in combination in patients with advanced carcinoids of the lung and thymus (LUNA): An open-label, multicentre, randomised, phase 2 trial. Lancet Oncol. 2017, 18, 1652–1664. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.E.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Yao, J.C.; Pavel, M.; Lombard-Bohas, C.; Van Cutsem, E.; Voi, M.; Brandt, U.; He, W.; Chen, D.; Capdevila, J.; de Vries, E.G.E.; et al. Everolimus for the Treatment of Advanced Pancreatic Neuroendocrine Tumors: Overall Survival and Circulating Biomarkers From the Randomized, Phase III RADIANT-3 Study. J. Clin. Oncol. 2016, 34, 3906–3913. [Google Scholar] [CrossRef]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/results?term=everolimus&cond=Neuroendocrine+Tumors&Search=Apply&recrs=b&recrs=a&recrs=f&recrs=d&age_v=&gndr=&type=&rslt= (accessed on 28 January 2018).
- Salazar, R.; Garcia-Carbonero, R.; Libutti, S.K.; Hendifar, A.E.; Custodio, A.; Guimbaud, R.; Lombard-Bohas, C.; Ricci, S.; Klümpen, H.; Capdevila, J.; et al. Phase II Study of BEZ235 versus Everolimus in Patients with Mammalian Target of Rapamycin Inhibitor-Naïve Advanced Pancreatic Neuroendocrine Tumors. Oncologist 2017. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety of Everolimus and (STZ-5FU) Given One Upfront the Other upon Progression in Advanced pNET (SEQTOR). Available online: https://clinicaltrials.gov/ct2/show/NCT02246127 (accessed on 28 January 2018).
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Rugo, H.S.; O’Neil, B.H.; Santoro, A.; Schellens, J.H.M.; Cohen, R.B.; Doi, T.; Ott, P.A.; Pishvaian, M.J.; Puzanov, I.; et al. Pembrolizumab for patients with PD-L1–positive advanced carcinoid or pancreatic neuroendocrine tumors: Results from the KEYNOTE-028 study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, v142. [Google Scholar] [CrossRef]
- Study of Efficacy and Safety of PDR001 in Patients with Advanced or Metastatic, Well-Differentiated, Non-Functional Neuroendocrine Tumors of Pancreatic, Gastrointestinal (GI), or Thoracic Origin or Poorly-Differentiated Gastroenteropancreatic Neuroendocri. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02955069 (accessed on 28 January 2018).
- Pembrolizumab Combined with Itacitinib (INCB039110) and/or Pembrolizumab Combined with INCB050465 in Advanced Solid Tumors. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02646748 (accessed on 28 January 2018).


{kind=link}
{kind=link}
| Title (Phase) | Year | Population | Treatments | PFS (HR) | Remarks |
|---|---|---|---|---|---|
| RADIANT-1 (II) | 2010 | 160 panNET | (I) Everolimus (II) Everolimus + Octreotide LAR | 9.7 16.7 | No comparison between strata |
| RADIANT-2 (III) | 2011 | 429 mixed (carcinoid syndrome) | Everolimus + Octreotide LAR vs. pbo + Octrotide LAR | 16.4 vs. 11.3 (HR: 0.77) | Not significant by central radiology analysis |
| RADIANT-3 (III) | 2011 | 410 panNET | Everolimus vs. pbo | 11 vs. 4.6 (HR: 0.35) | 40% concomitant SSA |
| RADIANT-4 (III) | 2016 | 302 mixed non-panceratic | Everolimus vs. pbo | 11 vs. 3.9 (HR: 0.48) | Concomitant SSA not allowed |
| LUNA trial (II) | 2017 | 124 thoracic (lung thymic) | Pasireotide Everolimus Everolimus + Pasireotide | 8.5 12.5 11.8 | No comparison among arms |
| Study | NCT | Study Design | Treatment |
|---|---|---|---|
| SEQTOR | NCT02246127 | Phase III, well differentiated panNET | Everolimus → STZ+5-FU vs. STZ+5-FU → Everlimus |
| Keynote-028 | NCT02054806 | Phase I, PD-L1-positive well-differentiated NETs | Pembrolizumab |
| CPDR001E2201 | NCT02955069 | Phase II, well-differentiated unselected thoracic, pancreatic and gastrointestinal NETs and GEP-NECs | PDR001 (anti PD-1) |
| 39110-107 | NCT02646748 | Phase I, advanced solid tumors | Pembrolizumab + INCB050465 (PI3K-delta inhibitor) (Group B) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamberti, G.; Brighi, N.; Maggio, I.; Manuzzi, L.; Peterle, C.; Ambrosini, V.; Ricci, C.; Casadei, R.; Campana, D. The Role of mTOR in Neuroendocrine Tumors: Future Cornerstone of a Winning Strategy? Int. J. Mol. Sci. 2018, 19, 747. https://doi.org/10.3390/ijms19030747
Lamberti G, Brighi N, Maggio I, Manuzzi L, Peterle C, Ambrosini V, Ricci C, Casadei R, Campana D. The Role of mTOR in Neuroendocrine Tumors: Future Cornerstone of a Winning Strategy? International Journal of Molecular Sciences. 2018; 19(3):747. https://doi.org/10.3390/ijms19030747
Chicago/Turabian StyleLamberti, Giuseppe, Nicole Brighi, Ilaria Maggio, Lisa Manuzzi, Chiara Peterle, Valentina Ambrosini, Claudio Ricci, Riccardo Casadei, and Davide Campana. 2018. "The Role of mTOR in Neuroendocrine Tumors: Future Cornerstone of a Winning Strategy?" International Journal of Molecular Sciences 19, no. 3: 747. https://doi.org/10.3390/ijms19030747
APA StyleLamberti, G., Brighi, N., Maggio, I., Manuzzi, L., Peterle, C., Ambrosini, V., Ricci, C., Casadei, R., & Campana, D. (2018). The Role of mTOR in Neuroendocrine Tumors: Future Cornerstone of a Winning Strategy? International Journal of Molecular Sciences, 19(3), 747. https://doi.org/10.3390/ijms19030747