The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
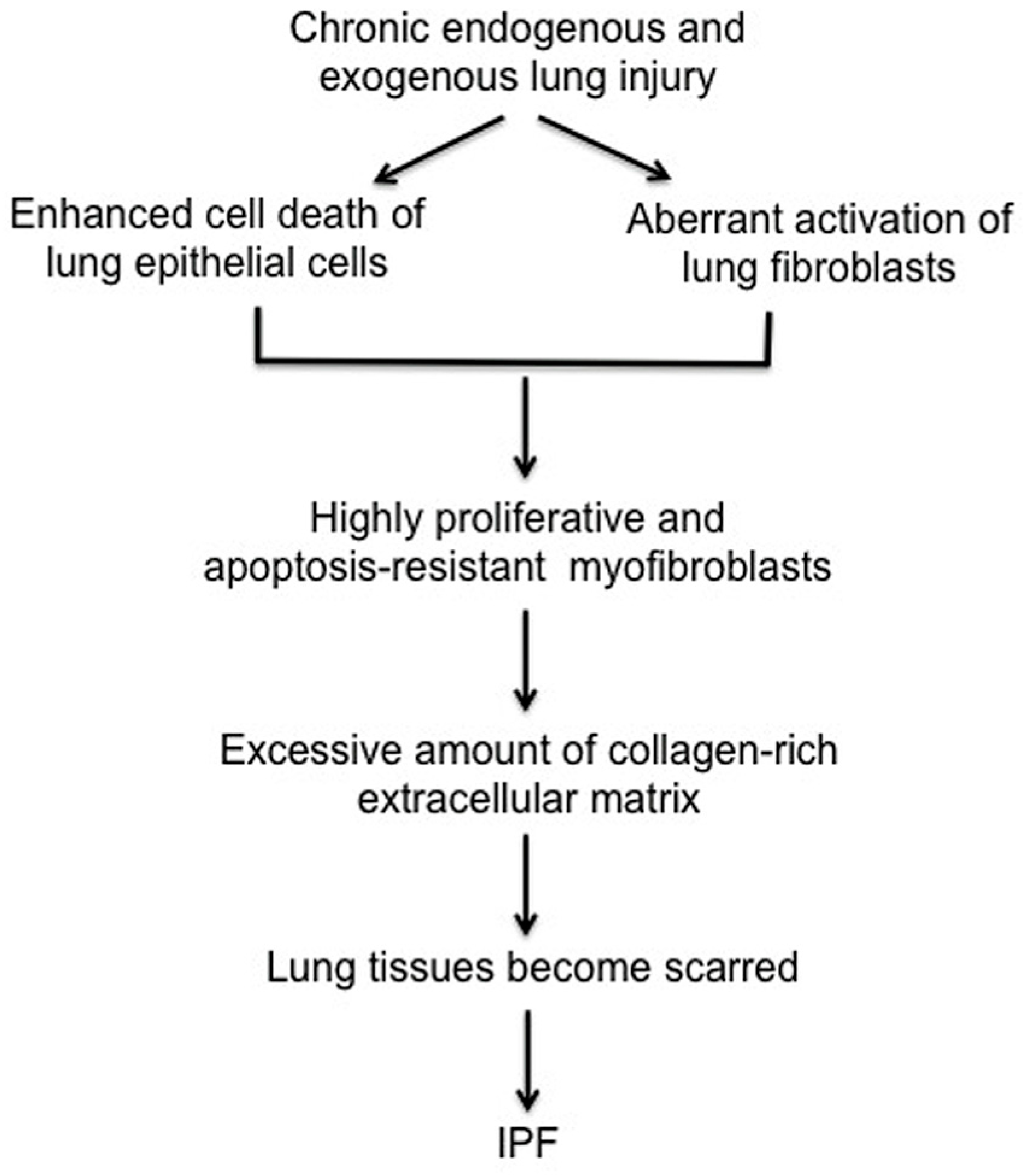
1.1. The Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF)
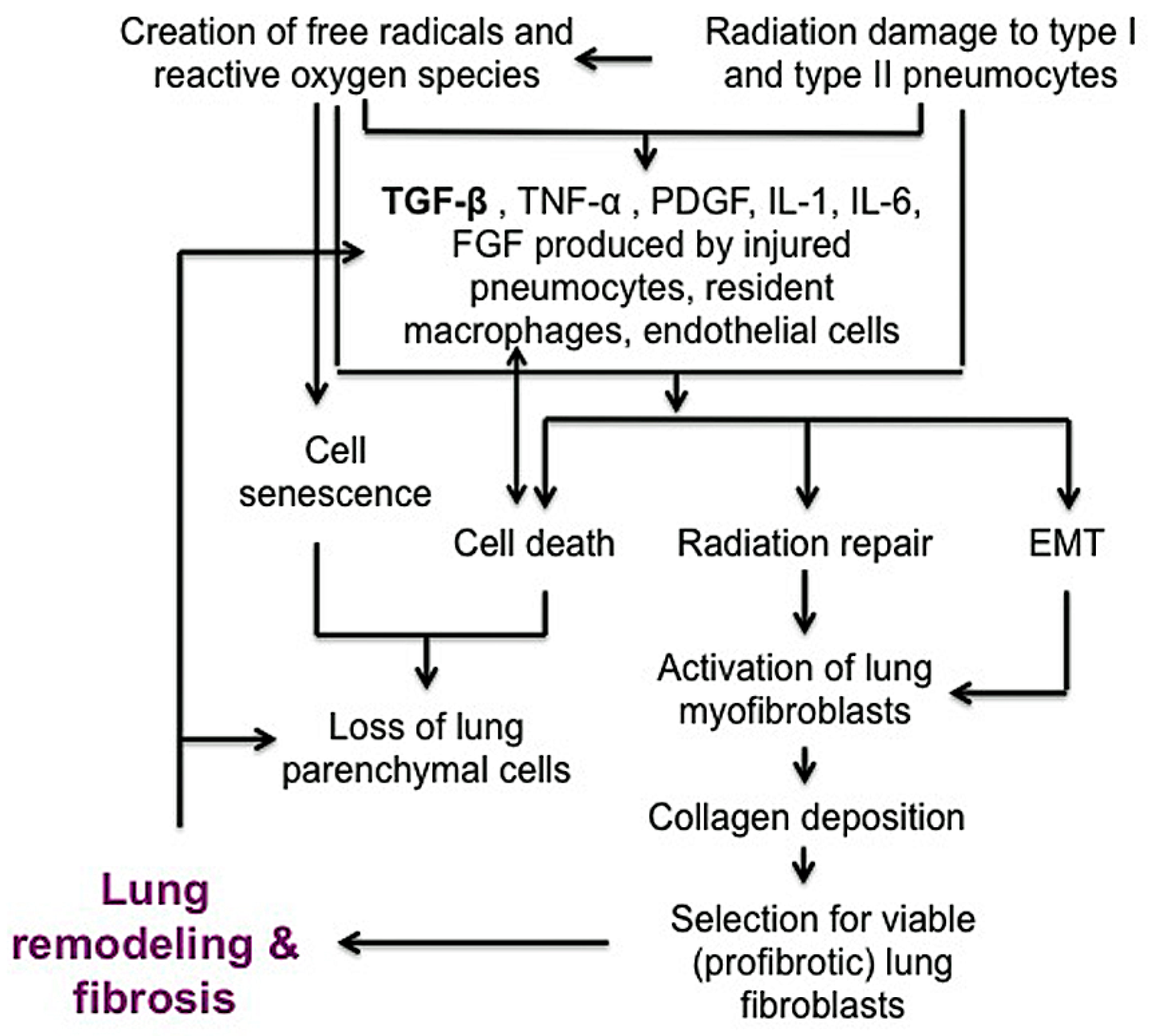
1.2. The Pathogenesis of Radiation-Induced Pulmonary Injury (RIPF)
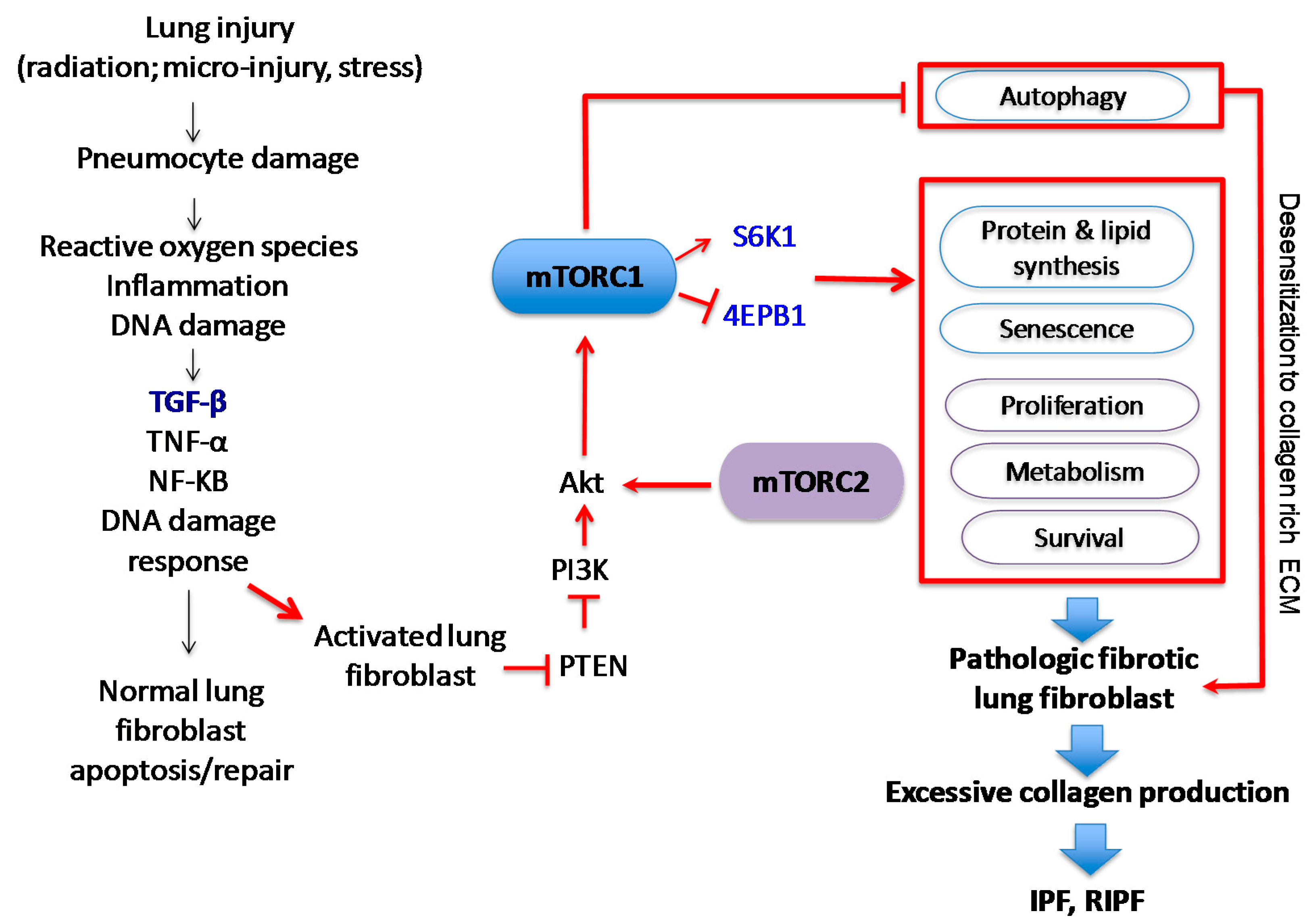
1.3. The Role of mTOR in Pulmonary Fibrosis
2. mTOR
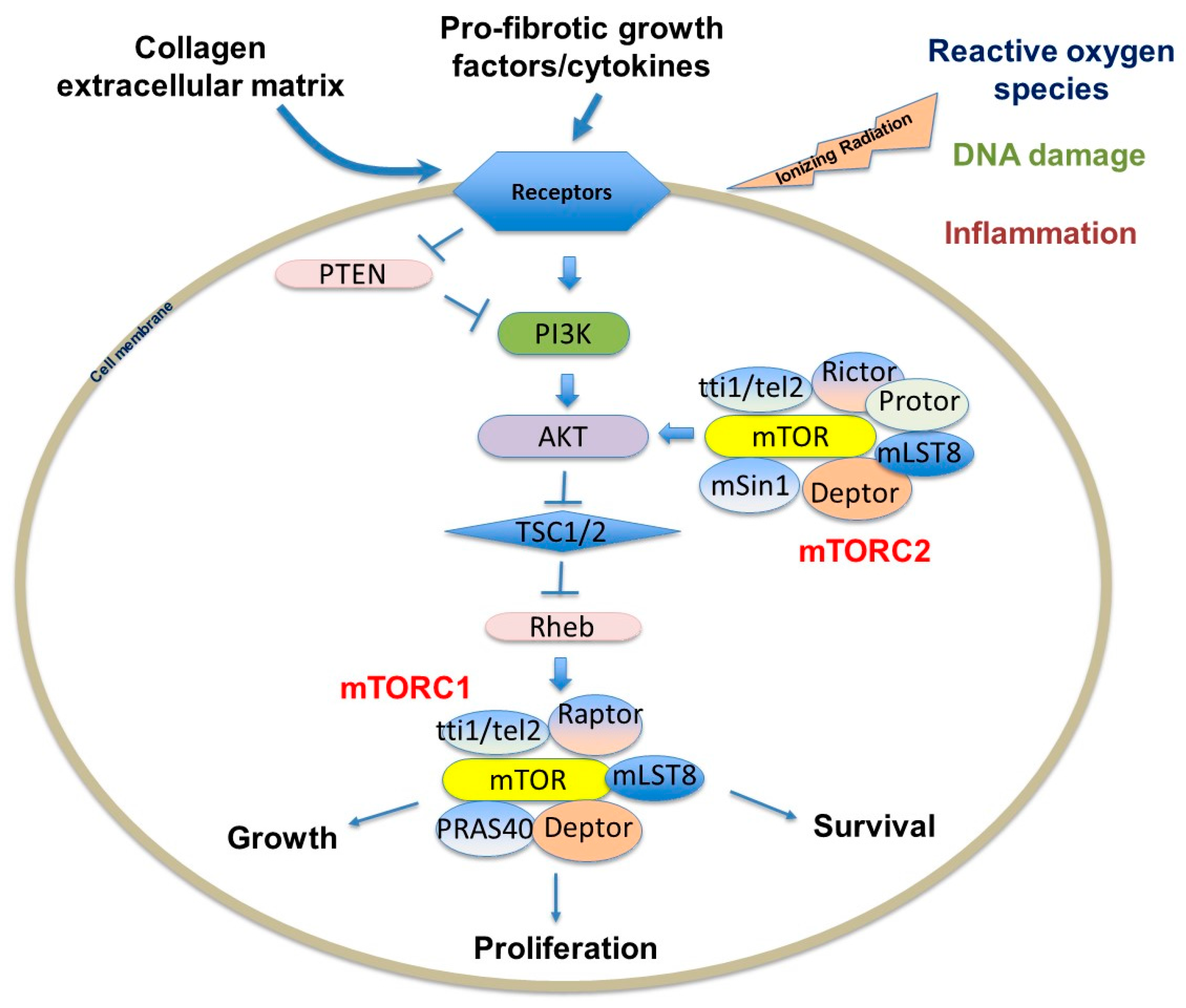
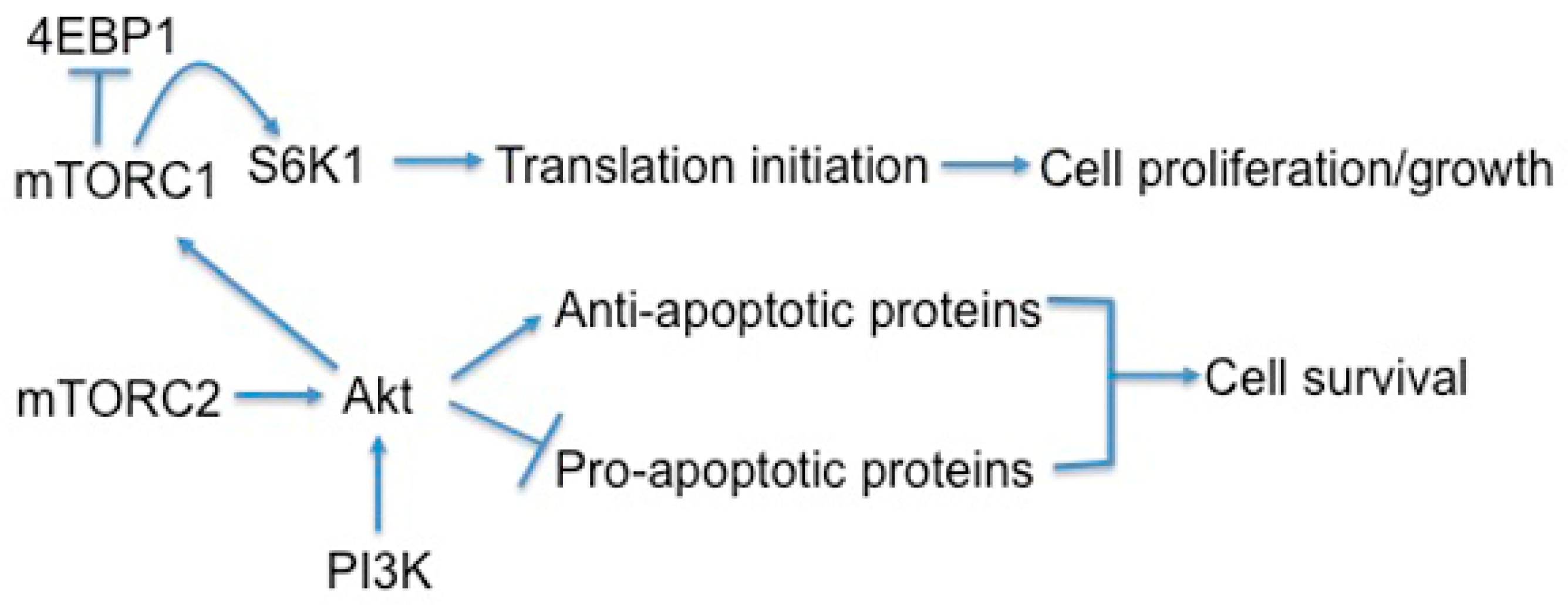
2.1. The Structure and Function of mTORC1
2.2. The Structure and Function of mTORC2
3. mTOR-Dependent Molecular Mechanisms that Promote Pulmonary Fibrosis
3.1. mTOR Regulates Cell Growth, Proliferation, and Viability
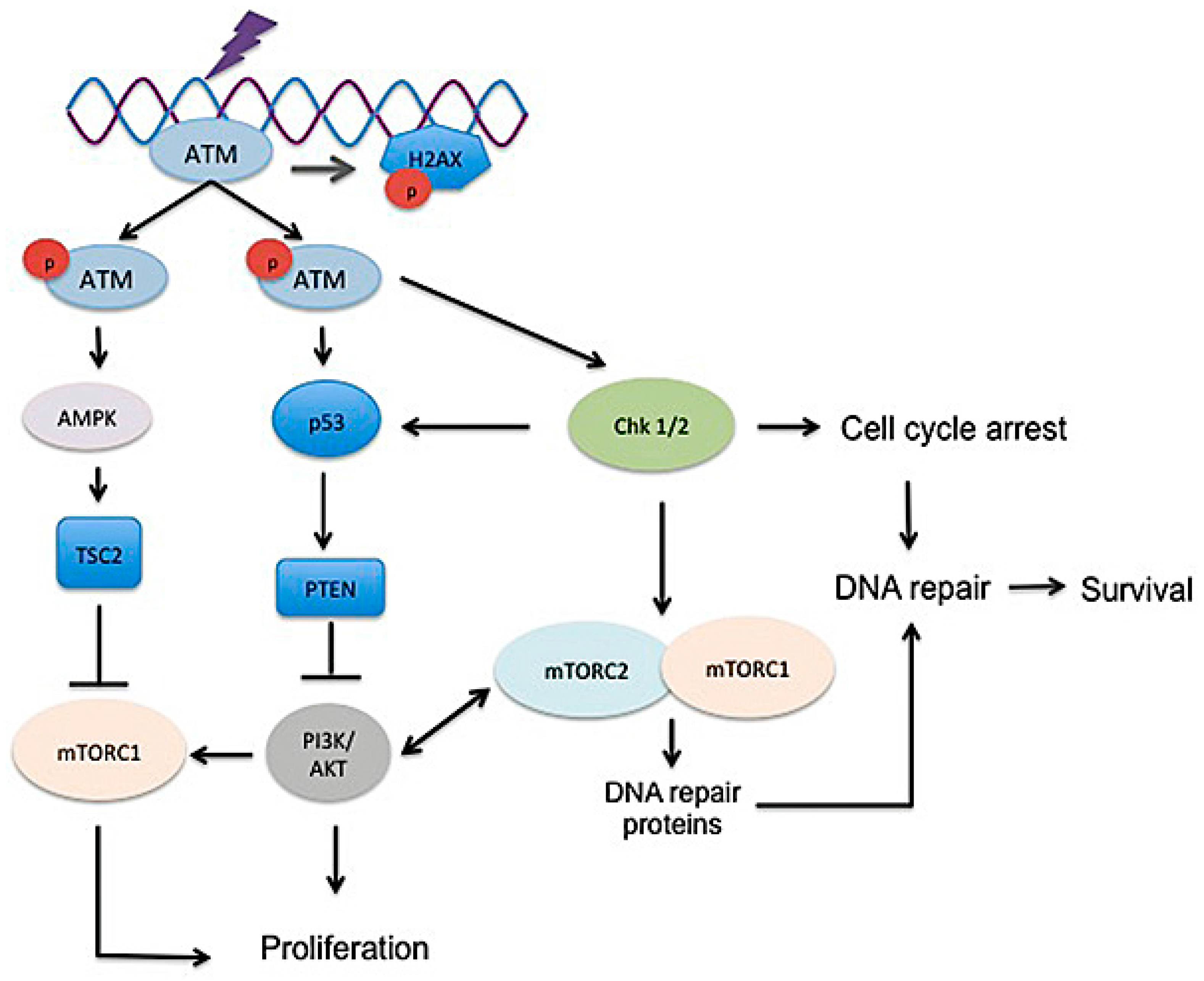
3.2. DNA Damage, Radiosensitivity, and DNA Damage Response
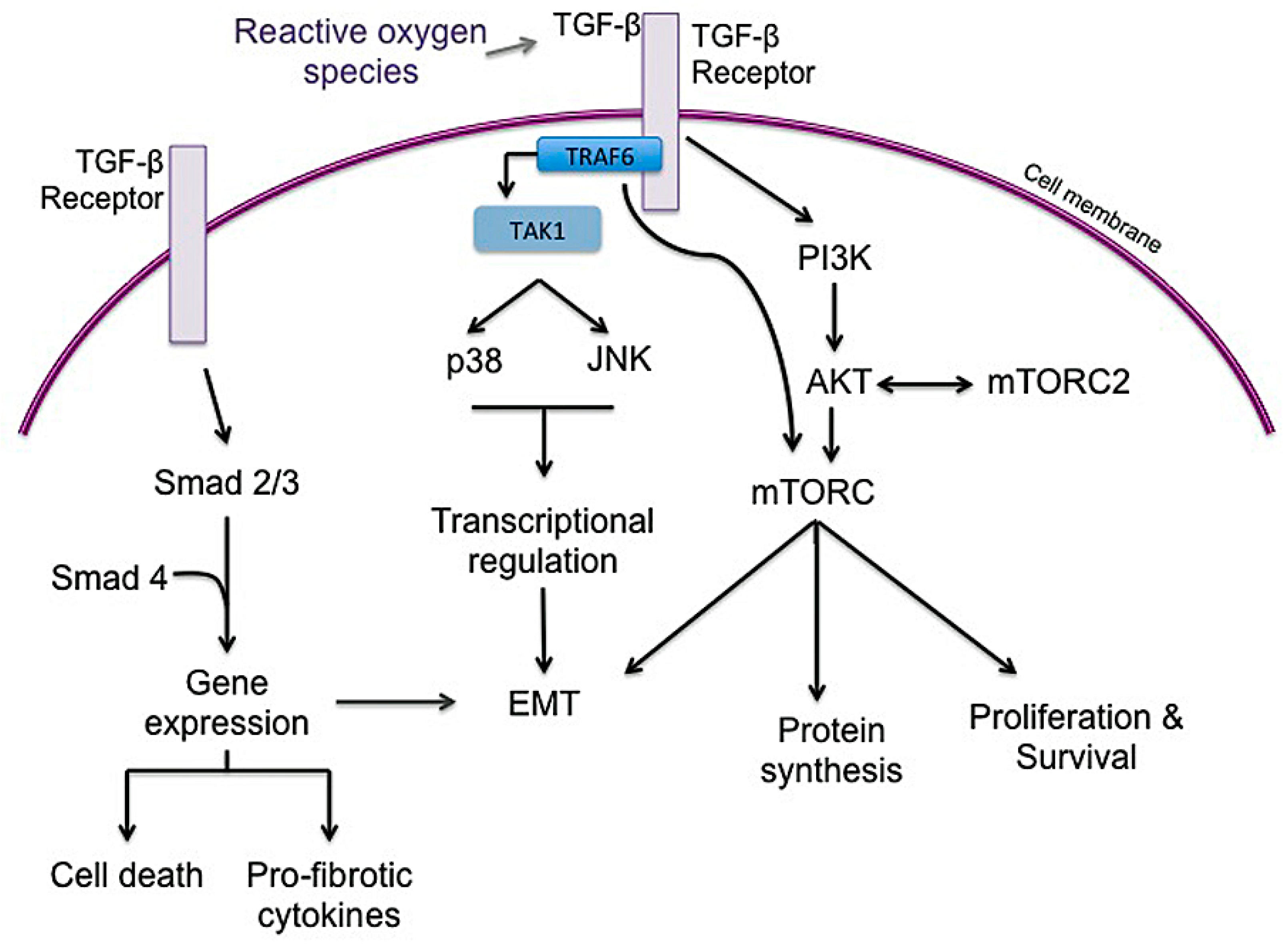
3.3. Inflammation
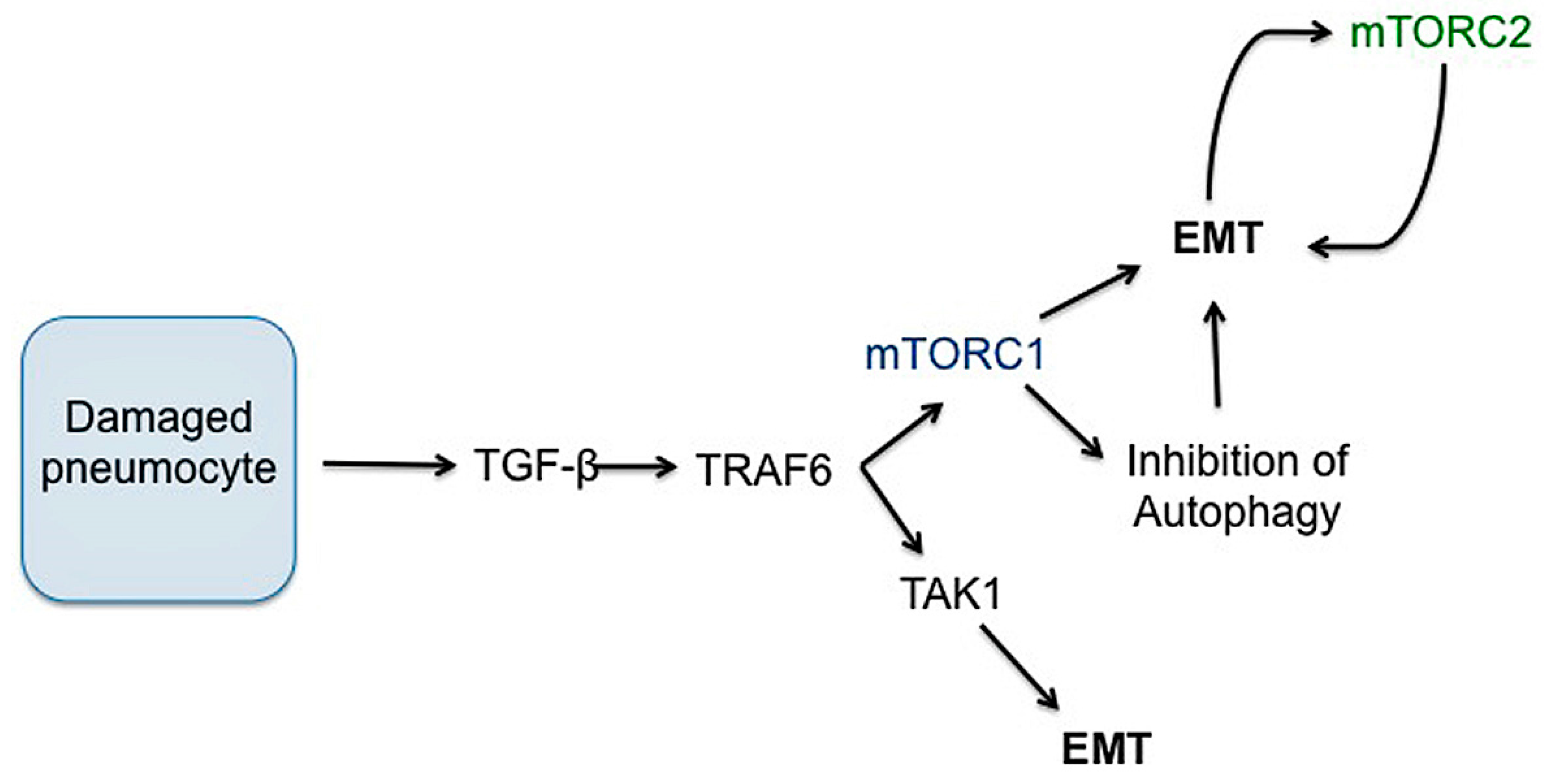
3.4. Epithelial to Mesenchymal Transition (EMT)
3.5. Autophagy
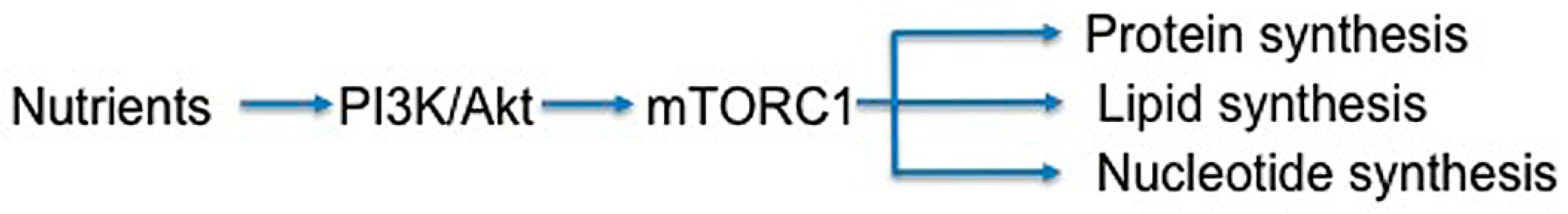
3.6. Metabolism
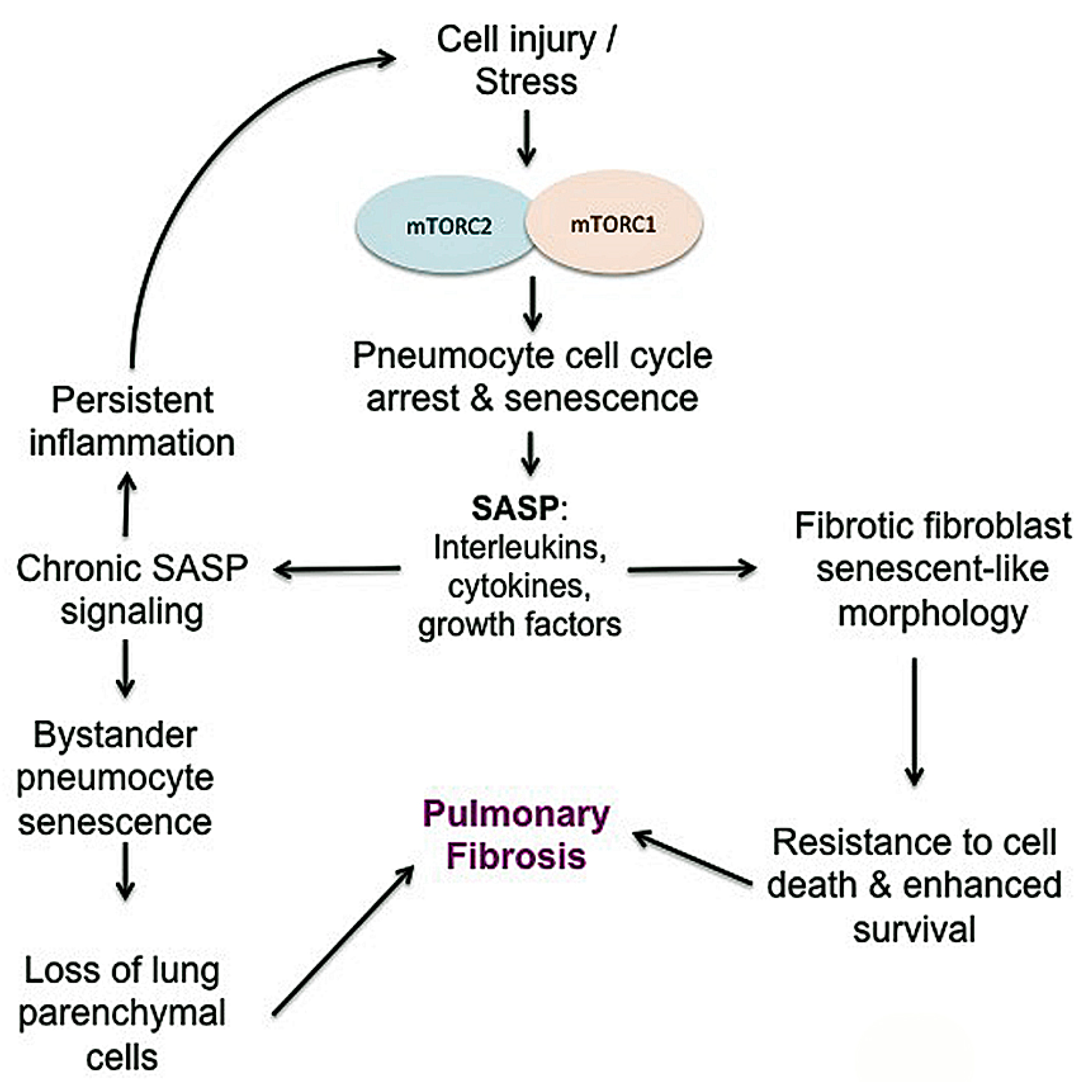
3.7. Senescence
4. Therapeutic Targeting of mTOR in Pulmonary Fibrosis
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Nanchahal, J.; Hinz, B. Strategies to overcome the hurdles to treat fibrosis, a major unmet clinical need. Proc. Natl. Acad. Sci. USA 2016, 113, 7291–7293. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, J.; Macarak, E.; Piera-Velazquez, S.; Jimenez, S.A. Human fibrotic diseases: Current challenges in fibrosis research. Methods Mol. Biol. 2017, 1627, 1–23. [Google Scholar] [PubMed]
- Antoniou, K.M.; Tomassetti, S.; Tsitoura, E.; Vancheri, C. Idiopathic pulmonary fibrosis and lung cancer: A clinical and pathogenesis update. Curr. Opin. Pulm. Med. 2015, 21, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Calio, A.; Lever, V.; Rossi, A.; Gilioli, E.; Brunelli, M.; Dubini, A.; Tomassetti, S.; Piciucchi, S.; Nottegar, A.; Rossi, G.; et al. Increased frequency of bronchiolar histotypes in lung carcinomas associated with idiopathic pulmonary fibrosis. Histopathology 2017, 71, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Karampitsakos, T.; Tzilas, V.; Tringidou, R.; Steiropoulos, P.; Aidinis, V.; Papiris, S.A.; Bouros, D.; Tzouvelekis, A. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, M.; Li, P.; Su, Z.; Gao, P.; Zhang, J. Idiopathic pulmonary fibrosis will increase the risk of lung cancer. Chin. Med. J. (Engl.) 2014, 127, 3142–3149. [Google Scholar] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- American Thoracic Society; European Respiratory Society. American thoracic society/european respiratory society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. This joint statement of the american thoracic society (ATS), and the european respiratory society (ERS) was adopted by the ats board of directors, June 2001 and by the ers executive committee, June 2001. Am. J. Respir. Crit. Care Med. 2002, 165, 277–304. [Google Scholar]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 2006, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the muc5b promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Gulati, M.; Redlich, C.A. Asbestosis and environmental causes of usual interstitial pneumonia. Curr. Opin. Pulm. Med. 2015, 21, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Phan, S.H. Inhibition of myofibroblast apoptosis by transforming growth factor β1. Am. J. Respir. Cell Mol. Biol. 1999, 21, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, G.A.; Sweeney, S.M.; Korkko, J.; Ala-Kokko, L.; San Antonio, J.D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type i collagen. J. Biol. Chem. 2002, 277, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, B. Rna protein interactions governing expression of the most abundant protein in human body, type i collagen. Wiley Interdiscip. Rev. RNA 2013, 4, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Danen, E.H.; Takino, T.; Miyamoto, S.; Yamada, K.M. Pten interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/AKT cell survival pathway. J. Biol. Chem. 1999, 274, 20693–20703. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Hergert, P.; Kahm, J.; Jessurun, J.; Henke, C. Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type i collagen matrix. Am. J. Pathol. 2011, 179, 2420–2430. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Diebold, D.; Nho, R.; Perlman, D.; Kleidon, J.; Kahm, J.; Avdulov, S.; Peterson, M.; Nerva, J.; Bitterman, P.; et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J. Exp. Med. 2008, 205, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Khalil, W.; Kahm, J.; Jessurun, J.; Kleidon, J.; Henke, C.A. Pathologic caveolin-1 regulation of pten in idiopathic pulmonary fibrosis. Am. J. Pathol. 2010, 176, 2626–2637. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Peterson, M.; Hergert, P.; Henke, C.A. FoxO3a (forkhead box O3a) deficiency protects idiopathic pulmonary fibrosis (ipf) fibroblasts from type i polymerized collagen matrix-induced apoptosis via caveolin-1 (cav-1) and fas. PLoS ONE 2013, 8, e61017. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr. Progress in cancer management. Keynote address. Cancer 1983, 51, 2401–2409. [Google Scholar] [CrossRef]
- Kong, F.M.; Hayman, J.A.; Griffith, K.A.; Kalemkerian, G.P.; Arenberg, D.; Lyons, S.; Turrisi, A.; Lichter, A.; Fraass, B.; Eisbruch, A.; et al. Final toxicity results of a radiation-dose escalation study in patients with non-small-cell lung cancer (nsclc): Predictors for radiation pneumonitis and fibrosis. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, M.H.; Cai, X.W.; Shedden, K.; Hayman, J.A.; Yuan, S.; Ritter, T.; Ten Haken, R.K.; Lawrence, T.S.; Kong, F.M. Combining physical and biologic parameters to predict radiation-induced lung toxicity in patients with non-small-cell lung cancer treated with definitive radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e217–e222. [Google Scholar] [CrossRef] [PubMed]
- Barriger, R.B.; Forquer, J.A.; Brabham, J.G.; Andolino, D.L.; Shapiro, R.H.; Henderson, M.A.; Johnstone, P.A.; Fakiris, A.J. A dose-volume analysis of radiation pneumonitis in non-small cell lung cancer patients treated with stereotactic body radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Ricardi, U.; Filippi, A.R.; Guarneri, A.; Giglioli, F.R.; Mantovani, C.; Fiandra, C.; Anglesio, S.; Ragona, R. Dosimetric predictors of radiation-induced lung injury in stereotactic body radiation therapy. Acta Oncol. 2009, 48, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Trovo, M.; Minatel, E.; Durofil, E.; Polesel, J.; Avanzo, M.; Baresic, T.; Bearz, A.; Del Conte, A.; Franchin, G.; Gobitti, C.; et al. Stereotactic body radiation therapy for re-irradiation of persistent or recurrent non-small cell lung cancer. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Ueki, N.; Matsuo, Y.; Togashi, Y.; Kubo, T.; Shibuya, K.; Iizuka, Y.; Mizowaki, T.; Togashi, K.; Mishima, M.; Hiraoka, M. Impact of pretreatment interstitial lung disease on radiation pneumonitis and survival after stereotactic body radiation therapy for lung cancer. J. Thorac. Oncol. 2015, 10, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Kunieda, E.; Takeda, T.; Tanaka, M.; Sanuki, N.; Fujii, H.; Shigematsu, N.; Kubo, A. Possible misinterpretation of demarcated solid patterns of radiation fibrosis on ct scans as tumor recurrence in patients receiving hypofractionated stereotactic radiotherapy for lung cancer. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Nakagawa, K.; Nakamura, N.; Koyanagi, H.; Tago, M.; Igaki, H.; Shiraishi, K.; Sasano, N.; Ohtomo, K. Exceptionally high incidence of symptomatic grade 2–5 radiation pneumonitis after stereotactic radiation therapy for lung tumors. Radiat. Oncol. 2007, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.R.; Siddiqui, F.; Anscher, M.S.; Movsas, B. Radiation pulmonary toxicity: From mechanisms to management. Semin. Radiat. Oncol. 2010, 20, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.P.; Johnston, C.J.; Finkelstein, J.N. Treatment for radiation-induced pulmonary late effects: Spoiled for choice or looking in the wrong direction? Curr. Drug Targets 2010, 11, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Brush, J.; Lipnick, S.L.; Phillips, T.; Sitko, J.; McDonald, J.T.; McBride, W.H. Molecular mechanisms of late normal tissue injury. Semin. Radiat. Oncol. 2007, 17, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Abratt, R.P.; Morgan, G.W. Lung toxicity following chest irradiation in patients with lung cancer. Lung Cancer 2002, 35, 103–109. [Google Scholar] [CrossRef]
- Zhang, X.J.; Sun, J.G.; Sun, J.; Ming, H.; Wang, X.X.; Wu, L.; Chen, Z.T. Prediction of radiation pneumonitis in lung cancer patients: A systematic review. J. Cancer Res. Clin. Oncol. 2012, 138, 2103–2116. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.; Lock, M.; D’Souza, D.; Yu, E.; Van Dyk, J. Prediction of radiation pneumonitis by dose-volume histogram parameters in lung cancer—A systematic review. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2004, 71, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, K.; Hashimoto, T.; Shimada, T.; Yoden, E.; Fujii, O.; Ota, Y.; Satouchi, M.; Negoro, S.; Adachi, S.; Soejima, T. Combined analysis of v20, vs5, pulmonary fibrosis score on baseline computed tomography, and patient age improves prediction of severe radiation pneumonitis after concurrent chemoradiotherapy for locally advanced non-small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Abe, T.; Omae, M.; Matsui, T.; Kato, M.; Hasegawa, H.; Enomoto, Y.; Ishihara, T.; Inui, N.; Yamada, K.; et al. Impact of preexisting interstitial lung disease on acute, extensive radiation pneumonitis: Retrospective analysis of patients with lung cancer. PLoS ONE 2015, 10, e0140437. [Google Scholar] [CrossRef] [PubMed]
- Mehta, V. Radiation pneumonitis and pulmonary fibrosis in non-small-cell lung cancer: Pulmonary function, prediction, and prevention. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Citrin, D.E.; Prasanna, P.G.S.; Walker, A.J.; Freeman, M.L.; Eke, I.; Barcellos-Hoff, M.H.; Arankalayil, M.J.; Cohen, E.P.; Wilkins, R.C.; Ahmed, M.M.; et al. Radiation-induced fibrosis: Mechanisms and opportunities to mitigate. Report of an nci workshop, september 19, 2016. Radiat. Res. 2017, 188, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.W.; Breit, S.N. Radiation and the lung: A reevaluation of the mechanisms mediating pulmonary injury. Int. J. Radiat. Oncol. Biol. Phys. 1995, 31, 361–369. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Lecarpentier, Y.; Guillevin, R.; Vallee, J.N. Interactions between TGF-beta1, canonical WNT/beta-catenin pathway and ppar gamma in radiation-induced fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef] [PubMed]
- Rube, C.E.; Uthe, D.; Schmid, K.W.; Richter, K.D.; Wessel, J.; Schuck, A.; Willich, N.; Rube, C. Dose-dependent induction of transforming growth factor beta (Tgf-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2000, 47, 1033–1042. [Google Scholar] [CrossRef]
- Almeida, C.; Nagarajan, D.; Tian, J.; Leal, S.W.; Wheeler, K.; Munley, M.; Blackstock, W.; Zhao, W. The role of alveolar epithelium in radiation-induced lung injury. PLoS ONE 2013, 8, e53628. [Google Scholar] [CrossRef] [PubMed]
- Rubin, P.; Johnston, C.J.; Williams, J.P.; McDonald, S.; Finkelstein, J.N. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 99–109. [Google Scholar] [CrossRef]
- Citrin, D.E.; Mitchell, J.B. Mechanisms of normal tissue injury from irradiation. Semin. Radiat. Oncol. 2017, 27, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Yan, H.; Li, L.; Yin, K.; Ji, F.; Zhang, S. Collagen triple helix repeat containing 1 (CTHRC1) activates integrin beta3/FAK signaling and promotes metastasis in ovarian cancer. J. Ovarian Res. 2017, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Sainson, R.C. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin. Cancer Biol. 2014, 25, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer associated fibroblasts: The architects of stroma remodelling. Proteomics 2017. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Polanska, U.M.; Orimo, A. Carcinoma-associated fibroblasts: Non-neoplastic tumour-promoting mesenchymal cells. J. Cell. Physiol. 2013, 228, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Morgan, R.N.; Adams, B.R.; Golding, S.E.; Dever, S.M.; Rosenberg, E.; Povirk, L.F.; Valerie, K. ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks. Cell Cycle 2011, 10, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Valerie, K.; Povirk, L.F. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003, 22, 5792–5812. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. Atm and atr substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. The PI3K/AKT/PTEN/mTOR pathway: A fruitful target for inducing cell death in rheumatoid arthritis? Future Med. Chem. 2015, 7, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Hennessy, B.T.; Slingerland, J.M. Next-generation mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J. Clin. Investig. 2011, 121, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.J.; Sowers, A.; Thetford, A.; McKay-Corkum, G.; Chung, S.I.; Mitchell, J.B.; Citrin, D.E. Mammalian target of rapamycin inhibition with rapamycin mitigates radiation-induced pulmonary fibrosis in a murine model. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Citrin, D.E.; Shankavaram, U.; Horton, J.A.; Shield, W., 3rd; Zhao, S.; Asano, H.; White, A.; Sowers, A.; Thetford, A.; Chung, E.J. Role of type ii pneumocyte senescence in radiation-induced lung fibrosis. J. Natl. Cancer Inst. 2013, 105, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.A.; Andrianifahanana, M.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Blenis, J.; Leof, E.B. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-beta. Cancer Res. 2009, 69, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-fkbp specifically blocks growth-dependent activation of and signaling by the 70 kd s6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Kuo, C.J.; Chung, J.; Fiorentino, D.F.; Flanagan, W.M.; Blenis, J.; Crabtree, G.R. Rapamycin selectively inhibits interleukin-2 activation of p70 s6 kinase. Nature 1992, 358, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liao, Y.; Xu, X.; Yi, Q.; He, L.; Tang, L. HnRNP A1 promotes keratinocyte cell survival post UVB radiation through PI3K/AKT/mTOR pathway. Exp. Cell Res. 2017, 362, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and AKT/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. A complex interplay between AKT, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, Y.; Zhou, X.; Qian, J.; Zhu, W.; Shu, Y.; Liu, P. Clinical efficacy of mTOR inhibitors in solid tumors: A systematic review. Future Oncol. 2015, 11, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Lazo, J.S.; Sharlow, E.R.; Epperly, M.W.; Lira, A.; Leimgruber, S.; Skoda, E.M.; Wipf, P.; Greenberger, J.S. Pharmacologic profiling of phosphoinositide 3-kinase inhibitors as mitigators of ionizing radiation-induced cell death. J. Pharmacol. Exp. Ther. 2013, 347, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Reale, A.; Marasco, C.; Vacca, A.; Carratu, M.R. Therapeutic targeting of the mTOR-signalling pathway in cancer: Benefits and limitations. Br. J. Pharmacol. 2014, 171, 3801–3813. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.M.; Belloli, E.A.; Stuckey, L.; Chan, K.M.; Lin, J.; Lynch, W.; Chang, A.; Mazzoni, S.M.; Fingar, D.C.; Lama, V.N. Mechanistic target of rapamycin Complex 1 (mTORC1) and mTORC2 as key signaling intermediates in mesenchymal cell activation. J. Biol. Chem. 2016, 291, 6262–6271. [Google Scholar] [CrossRef] [PubMed]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 s6 kinase and 4E-BP1 through their tor signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. Tos motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. Mtor kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. Deptor is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. Pras40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. Pras40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.A.; Leite, F.G.; Andrade, L.G.; Torres, A.A.; De Sousa, L.P.; Barcelos, L.S.; Teixeira, M.M.; Ferreira, P.C.; Kroon, E.G.; Souto-Padron, T.; et al. Activation of the PI3K/AKT pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J. Virol. 2009, 83, 6883–6899. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Wei, K.; Ho, L.; Berry, G.J.; Jacobs, S.S.; Chang, C.H.; Rosen, G.D. A critical role for the mTORC2 pathway in lung fibrosis. PLoS ONE 2014, 9, e106155. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villen, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin—PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Shah, O.J.; Wang, Z.; Hunter, T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 2004, 14, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Nasmyth, K. Retinoblastoma protein. Another role rolls in. Nature 1996, 382, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Cope, C.L.; Gilley, R.; Balmanno, K.; Sale, M.J.; Howarth, K.D.; Hampson, M.; Smith, P.D.; Guichard, S.M.; Cook, S.J. Adaptation to mTOR kinase inhibitors by amplification of eif4e to maintain cap-dependent translation. J. Cell Sci. 2014, 127, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rivera, E.; Jayaraman, P.; Parikh, F.; Davies, M.A.; Ekmekcioglu, S.; Izadmehr, S.; Milton, D.R.; Chipuk, J.E.; Grimm, E.A.; Estrada, Y.; et al. Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of tsc2. Cancer Res. 2014, 74, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Haendeler, J.; Goebel, C.; Zeiher, A.M.; Dimmeler, S. Growth factor-induced phosphoinositide 3-OH kinase/AKT phosphorylation in smooth muscle cells: Induction of cell proliferation and inhibition of cell death. Cardiovasc. Res. 2000, 48, 148–157. [Google Scholar] [CrossRef]
- Shelton, J.G.; Moye, P.W.; Steelman, L.S.; Blalock, W.L.; Lee, J.T.; Franklin, R.A.; McMahon, M.; McCubrey, J.A. Differential effects of kinase cascade inhibitors on neoplastic and cytokine-mediated cell proliferation. Leukemia 2003, 17, 1765–1782. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Flynn, D.C.; Zhang, Z.; Zhong, X.S.; Walker, V.; Liu, K.J.; Shi, X.; Jiang, B.H. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70s6k1 signaling in human ovarian cancer cells. Am. J. Physiol. Cell Physiol. 2004, 287, C281–C291. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.G.; Blalock, W.L.; White, E.R.; Steelman, L.S.; McCubrey, J.A. Ability of the activated PI3K/AKT oncoproteins to synergize with MEK1 and induce cell cycle progression and abrogate the cytokine-dependence of hematopoietic cells. Cell Cycle 2004, 3, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Bergholz, U.; Jucker, M.; McCubrey, J.A.; Trumper, L.; Stocking, C.; Basecke, J. Mutations in the catalytic subunit of class ia PI3K confer leukemogenic potential to hematopoietic cells. Oncogene 2008, 27, 4096–4106. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Kang, S.; Zhao, L.; Vogt, P.K. Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 2005, 5, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Parker, V.E.; Knox, R.G.; Zhang, Q.; Wakelam, M.J.; Semple, R.K. Phosphoinositide 3-kinase-related overgrowth: Cellular phenotype and future therapeutic options. Lancet 2015, 385 (Suppl. S1). [Google Scholar] [CrossRef]
- Das, F.; Ghosh-Choudhury, N.; Mariappan, M.M.; Kasinath, B.S.; Choudhury, G.G. Hydrophobic motif site-phosphorylated protein kinase cbetaii between mTORC2 and akt regulates high glucose-induced mesangial cell hypertrophy. Am. J. Physiol. Cell Physiol. 2016, 310, C583–C596. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.S.; Liu, C.C.; Lin, J.H.; Hsu, T.W.; Hsu, J.W.; Su, K.; Hung, S.C. Involvement of er stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 2017, 7, 14272. [Google Scholar] [CrossRef] [PubMed]
- Syed, F.; Sherris, D.; Paus, R.; Varmeh, S.; Singh, S.; Pandolfi, P.P.; Bayat, A. Keloid disease can be inhibited by antagonizing excessive mTOR signaling with a novel dual torc1/2 inhibitor. Am. J. Pathol. 2012, 181, 1642–1658. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of AKT/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 2015, 162, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Hergert, P. Ipf fibroblasts are desensitized to type i collagen matrix-induced cell death by suppressing low autophagy via aberrant AKT/mTOR kinases. PLoS ONE 2014, 9, e94616. [Google Scholar] [CrossRef] [PubMed]
- Strimpakos, A.S.; Karapanagiotou, E.M.; Saif, M.W.; Syrigos, K.N. The role of mTOR in the management of solid tumors: An overview. Cancer Treat. Rev. 2009, 35, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, L.; Liu, X.; Yuan, C.; Wang, G. Improved antitumor effect of ionizing radiation in combination with rapamycin for treating nasopharyngeal carcinoma. Oncol. Lett. 2017, 14, 1105–1108. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor tak228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Makinde, A.Y.; Aryankalayil, M.J.; Sandfort, V.; Palayoor, S.T.; Rath, B.H.; Liotta, L.; Pierobon, M.; Petricoin, E.F.; Brown, M.F.; et al. Exploiting radiation-induced signaling to increase the susceptibility of resistant cancer cells to targeted drugs: AKT and mTOR inhibitors as an example. Mol. Cancer Ther. 2017, 17, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Takahashi, A.; Ohnishi, K.; Ota, I.; Ohnishi, T.; Tojo, T.; Taniguchi, S. Effect of rapamycin, an mTOR inhibitor, on radiation sensitivity of lung cancer cells having different p53 gene status. Int. J. Oncol. 2010, 37, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Dumont, F.J.; Bischoff, P. Disrupting the mTOR signaling network as a potential strategy for the enhancement of cancer radiotherapy. Curr. Cancer Drug Targets 2012, 12, 899–924. [Google Scholar] [CrossRef] [PubMed]
- Sharlow, E.R.; Leimgruber, S.; Lira, A.; McConnell, M.J.; Norambuena, A.; Bloom, G.S.; Epperly, M.W.; Greenberger, J.S.; Lazo, J.S. A small molecule screen exposes mTOR signaling pathway involvement in radiation-induced apoptosis. ACS Chem. Biol. 2016, 11, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Patel, V.; Cotrim, A.; Leelahavanichkul, K.; Molinolo, A.A.; Mitchell, J.B.; Gutkind, J.S. Mtor inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell 2012, 11, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yamada, E.; Zong, H.; Pessin, J.E. Fyn activation of mTORC1 stimulates the ire1alpha-JNK pathway, leading to cell death. J. Biol. Chem. 2015, 290, 24772–24783. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, tsc2, and pten expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the igf-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Gillick, J.L.; Neil, J.; Tobias, M.; Thwing, Z.E.; Murali, R. Distinct signaling mechanisms of mTORC1 and mTORC2 in glioblastoma multiforme: A tale of two complexes. Adv. Biol. Regul. 2015, 57, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Hayman, T.J.; Wahba, A.; Rath, B.H.; Bae, H.; Kramp, T.; Shankavaram, U.T.; Camphausen, K.; Tofilon, P.J. The ATP-competitive mTOR inhibitor INK128 enhances in vitro and in vivo radiosensitivity of pancreatic carcinoma cells. Clin. Cancer Res. 2014, 20, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Tang, J.; Chen, Z.; Zhang, H.; Wang, H.; Yang, J.; Zhang, H. The novel mTORC1/2 dual inhibitor ink128 enhances radiosensitivity of breast cancer cell line MCF-7. Int. J. Oncol. 2016, 49, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Zellefrow, C.D.; Sharlow, E.R.; Epperly, M.W.; Reese, C.E.; Shun, T.; Lira, A.; Greenberger, J.S.; Lazo, J.S. Identification of druggable targets for radiation mitigation using a small interfering rna screening assay. Radiat. Res. 2012, 178, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Willers, H.; Held, K.D. Introduction to clinical radiation biology. Hematol. Oncol. Clin. N. Am. 2006, 20, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.; McMillan, T.J. DNA damage and repair following treatment with ionizing radiation. Radiother. Oncol. 1990, 19, 95–108. [Google Scholar] [CrossRef]
- Wang, W.H.; Chien, T.H.; Fan, S.M.; Huang, W.Y.; Lai, S.F.; Wu, J.T.; Lin, S.J. Activation of mTORC1 signaling is required for timely hair follicle regeneration from radiation injury. Radiat. Res. 2017, 188, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Nho, R.S.; Kahm, J.; Kleidon, J.; Henke, C.A. Focal adhesion kinase is upstream of phosphatidylinositol 3-Kinase/AKT in regulating fibroblast survival in response to contraction of type i collagen matrices via a beta 1 integrin viability signaling pathway. J. Biol. Chem. 2004, 279, 33024–33034. [Google Scholar] [CrossRef] [PubMed]
- Udayakumar, D.; Pandita, R.K.; Horikoshi, N.; Liu, Y.; Liu, Q.; Wong, K.K.; Hunt, C.R.; Gray, N.S.; Minna, J.D.; Pandita, T.K.; et al. Torin2 suppresses ionizing radiation-induced DNA damage repair. Radiat. Res. 2016, 185, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Beuvink, I.; Boulay, A.; Fumagalli, S.; Zilbermann, F.; Ruetz, S.; O’Reilly, T.; Natt, F.; Hall, J.; Lane, H.A.; Thomas, G. The mTOR inhibitor rad001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell 2005, 120, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, J.; Elia, A.; Carroll, V.A.; Moumen, A. DNA damage-induced s and g2/m cell cycle arrest requires mTORC2-dependent regulation of chk1. Oncotarget 2015, 6, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Albert, J.M.; Kim, K.W.; Cao, C.; Lu, B. Targeting the AKT/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol. Cancer Ther. 2006, 5, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Silvera, D.; Ernlund, A.; Arju, R.; Connolly, E.; Volta, V.; Wang, J.; Schneider, R.J. MTORC1 and -2 coordinate transcriptional and translational reprogramming in resistance to DNA damage and replicative stress in breast cancer cells. Mol. Cell. Biol. 2017, 37, e00577-16. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Lofroth, P.O.; Johansson, L.; Riklund, K.A.; Stigbrand, T. Cell cycle disturbances and mitotic catastrophes in HeLa Hep2 cells following 2.5 to 10 gy of ionizing radiation. Clin. Cancer Res. 2007, 13, 5501s–5508s. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Valent, A.; Raslova, H.; Yakushijin, K.; Horne, D.; Feunteun, J.; Lenoir, G.; Medema, R.; et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004, 23, 4362–4370. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Lofroth, P.O.; Johansson, L.; Riklund, K.; Stigbrand, T. Apoptotic signalling in HeLa Hep2 cells following 5 Gy of cobalt-60 gamma radiation. Anticancer Res. 2009, 29, 4361–4366. [Google Scholar] [PubMed]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologis, 7th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Im-aram, A.; Farrand, L.; Bae, S.M.; Song, G.; Song, Y.S.; Han, J.Y.; Tsang, B.K. The mTORC2 component rictor contributes to cisplatin resistance in human ovarian cancer cells. PLoS ONE 2013, 8, e75455. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zang, C.; Schefe, J.H.; Schwarzlose-Schwarck, S.; Regierer, A.C.; Elstner, E.; Schulz, C.O.; Scholz, C.; Possinger, K.; Eucker, J. The mTOR inhibitor rad001 sensitizes tumor cells to the cytotoxic effect of carboplatin in breast cancer in vitro. Anticancer Res. 2011, 31, 2713–2722. [Google Scholar] [PubMed]
- Liu, H.; Scholz, C.; Zang, C.; Schefe, J.H.; Habbel, P.; Regierer, A.C.; Schulz, C.O.; Possinger, K.; Eucker, J. Metformin and the mTOR inhibitor everolimus (rad001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro. Anticancer Res. 2012, 32, 1627–1637. [Google Scholar] [PubMed]
- Budanov, A.V.; Karin, M. P53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, J.; Du, W.; Zhang, S.; O’Connor, M.; Thomas, G.; Kozma, S.; Zingarelli, B.; Pang, Q.; Zheng, Y. Mtor regulates DNA damage response through nf-kappab-mediated FANCD2 pathway in hematopoietic cells. Leukemia 2013, 27, 2040–2046. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Oswald, D.; Phelps, D.; Cam, H.; Pelloski, C.E.; Pang, Q.; Houghton, P.J. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res. 2013, 73, 3393–3401. [Google Scholar] [CrossRef] [PubMed]
- Abaji, C.; Cousineau, I.; Belmaaza, A. Brca2 regulates homologous recombination in response to DNA damage: Implications for genome stability and carcinogenesis. Cancer Res. 2005, 65, 4117–4125. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Li, L.; Li, Y.; Wu, C.; Yin, Y.; Chen, Y.; Deng, M.; Nowsheen, S.; Yuan, J.; Lou, Z. A phosphorylation-deubiquitination cascade regulates the brca2-rad51 axis in homologous recombination. Genes Dev. 2016, 30, 2581–2595. [Google Scholar] [CrossRef] [PubMed]
- Nestal de Moraes, G.; Bella, L.; Zona, S.; Burton, M.J.; Lam, E.W. Insights into a critical role of the FOXO3a-FOXM1 axis in DNA damage response and genotoxic drug resistance. Curr. Drug Targets 2016, 17, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Raychaudhuri, P.; Costa, R.H. Chk2 mediates stabilization of the foxm1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell. Biol. 2007, 27, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Balli, D.; Ustiyan, V.; Zhang, Y.; Wang, I.C.; Masino, A.J.; Ren, X.; Whitsett, J.A.; Kalinichenko, V.V.; Kalin, T.V. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013, 32, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Karadedou, C.T.; Gomes, A.R.; Chen, J.; Petkovic, M.; Ho, K.K.; Zwolinska, A.K.; Feltes, A.; Wong, S.Y.; Chan, K.Y.; Cheung, Y.N.; et al. FoxO3a represses vegf expression through foxm1-dependent and -independent mechanisms in breast cancer. Oncogene 2012, 31, 1845–1858. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Im, J.; Ho, Y.Y.; Hergert, P. Microrna-96 inhibits FoxO3a function in ipf fibroblasts on type i collagen matrix. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L632–L642. [Google Scholar] [CrossRef] [PubMed]
- Im, J.; Hergert, P.; Nho, R.S. Reduced FoxO3a expression causes low autophagy in idiopathic pulmonary fibrosis fibroblasts on collagen matrices. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L552–L561. [Google Scholar] [CrossRef] [PubMed]
- Calveley, V.L.; Khan, M.A.; Yeung, I.W.; Vandyk, J.; Hill, R.P. Partial volume rat lung irradiation: Temporal fluctuations of in-field and out-of-field DNA damage and inflammatory cytokines following irradiation. Int. J. Radiat. Biol. 2005, 81, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Rube, C.E.; Wilfert, F.; Palm, J.; Konig, J.; Burdak-Rothkamm, S.; Liu, L.; Schuck, A.; Willich, N.; Rube, C. Irradiation induces a biphasic expression of pro-inflammatory cytokines in the lung. Strahlenther. Onkol. 2004, 180, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Herskind, C.; Bamberg, M.; Rodemann, H.P. The role of cytokines in the development of normal-tissue reactions after radiotherapy. Strahlenther. Onkol. 1998, 174 (Suppl. S3), 12–15. [Google Scholar] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Papiris, S.A.; Tomos, I.P.; Karakatsani, A.; Spathis, A.; Korbila, I.; Analitis, A.; Kolilekas, L.; Kagouridis, K.; Loukides, S.; Karakitsos, P.; et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine 2017, 102, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gonzalez, F.J.; Chandel, N.S.; Jain, M.; Budinger, G.R.S. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl. Res. 2017, 190, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Koli, K.; Myllarniemi, M.; Keski-Oja, J.; Kinnula, V.L. Transforming growth factor-beta activation in the lung: Focus on fibrosis and reactive oxygen species. Antioxid. Redox Signal. 2008, 10, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Dancea, H.C.; Shareef, M.M.; Ahmed, M.M. Role of radiation-induced TGF-beta signaling in cancer therapy. Mol. Cell. Pharmacol. 2009, 1, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the tgfbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Chen, X.; Wang, Q.; Wu, S.; Zheng, Y.; Liu, X. Role of the MAPKs/TGF-beta1/TRAF6 signaling pathway in postoperative atrial fibrillation. PLoS ONE 2017, 12, e0173759. [Google Scholar]
- Ji, Y.X.; Zhang, P.; Zhang, X.J.; Zhao, Y.C.; Deng, K.Q.; Jiang, X.; Wang, P.X.; Huang, Z.; Li, H. The ubiquitin e3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via tak1-dependent signalling. Nat. Commun. 2016, 7, 11267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, X.; Chen, X.; Gu, J.; Li, F.; Zhang, W.; Zheng, Y. Role of the MAPKs/TGF-beta1/TRAF6 signaling pathway in atrial fibrosis of patients with chronic atrial fibrillation and rheumatic mitral valve disease. Cardiology 2014, 129, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Divakaran, V.G.; Evans, S.; Topkara, V.K.; Diwan, A.; Burchfield, J.; Gao, F.; Dong, J.; Tzeng, H.P.; Sivasubramanian, N.; Barger, P.M.; et al. Tumor necrosis factor receptor-associated factor 2 signaling provokes adverse cardiac remodeling in the adult mammalian heart. Circ. Heart Fail. 2013, 6, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Deng, K.Q.; Luo, Y.; Jiang, D.S.; Gao, L.; Zhang, X.F.; Zhang, P.; Zhao, G.N.; Zhu, X.; Li, H. Tumor necrosis factor receptor-associated factor 3 is a positive regulator of pathological cardiac hypertrophy. Hypertension 2015, 66, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, D.; Zhang, X.; Jiang, M.; Hu, C.; Lin, J.; Tang, J.; Wu, L. Cardiac-specific TRAF2 overexpression enhances cardiac hypertrophy through activating AKT/GSK3BETA signaling. Gene 2014, 536, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Moon, G.; Kim, J.; Min, Y.; Wi, S.M.; Shim, J.H.; Chun, E.; Lee, K.Y. Phosphoinositide-dependent kinase-1 inhibits TRAF6 ubiquitination by interrupting the formation of TAK1-TAB2 complex in TLR4 signaling. Cell. Signal. 2015, 27, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates smad-independent activation of jnk and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Campbell, J.; Stenmark, M.H.; Zhao, J.; Stanton, P.; Matuszak, M.M.; Ten Haken, R.K.; Kong, F.S. Plasma levels of il-8 and TGF-beta1 predict radiation-induced lung toxicity in non-small cell lung cancer: A validation study. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, L.; Ji, W.; Wang, X.; Zhu, X.; Hayman, J.A.; Kalemkerian, G.P.; Yang, W.; Brenner, D.; Lawrence, T.S.; et al. Elevation of plasma TGF-beta1 during radiation therapy predicts radiation-induced lung toxicity in patients with non-small-cell lung cancer: A combined analysis from beijing and michigan. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.J.; Piedboeuf, B.; Rubin, P.; Williams, J.P.; Baggs, R.; Finkelstein, J.N. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumor necrosis factor alpha mrna levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat. Res. 1996, 145, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, G.; Qiao, T.; Yuan, S.; Zhuang, X. Effects of CpG oligodeoxynucleotide 1826 on acute radiation-induced lung injury in mice. Biol. Res. 2016, 49, 8. [Google Scholar] [CrossRef] [PubMed]
- Le, Z.; Niu, X.; Chen, Y.; Ou, X.; Zhao, G.; Liu, Q.; Tu, W.; Hu, C.; Kong, L.; Liu, Y. Predictive single nucleotide polymorphism markers for acute oral mucositis in patients with nasopharyngeal carcinoma treated with radiotherapy. Oncotarget 2017, 8, 63026–63037. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.P.; Zaidi, A.; Mahmood, J.; Jelveh, S. Investigations into the role of inflammation in normal tissue response to irradiation. Radiother. Oncol. 2011, 101, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.M. Regulation of radiation-induced apoptosis by early growth response-1 gene in solid tumors. Curr. Cancer Drug Targets 2004, 4, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, S.; Dey, S.; Meigooni, A.S.; Regine, W.F.; Kudrimoti, M.S.; Ahmed, M.M.; Mohiuddin, M. The impact of tnf-alpha induction on therapeutic efficacy following high dose spatially fractionated (grid) radiation. Technol. Cancer Res. Treat. 2002, 1, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Favaudon, V.; Caplier, L.; Monceau, V.; Pouzoulet, F.; Sayarath, M.; Fouillade, C.; Poupon, M.F.; Brito, I.; Hupe, P.; Bourhis, J.; et al. Ultrahigh dose-rate flash irradiation increases the differential response between normal and tumor tissue in mice. Sci. Transl. Med. 2014, 6, 245ra293. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Linke, M.; Pham, H.T.; Katholnig, K.; Schnoller, T.; Miller, A.; Demel, F.; Schutz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat. Immunol. 2017, 18, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Sunilgowda, S.N.; Nagarajan, D. Radiation-induced pulmonary epithelial-mesenchymal transition: A review of targeting molecular pathways and mediators. Curr. Drug Targets 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, L.F.; Kao, K.C.; Liu, Y.Y.; Lin, C.W.; Chen, N.H.; Lee, C.S.; Wang, C.W.; Yang, C.T. Nintedanib reduces ventilation-augmented bleomycin-induced epithelial-mesenchymal transition and lung fibrosis through suppression of the src pathway. J. Cell. Mol. Med. 2017, 21, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. Epithelial-mesenchymal interactions in fibrosis and repair. Transforming growth factor-beta activation by epithelial cells and fibroblasts. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S1), S21–S23. [Google Scholar] [CrossRef] [PubMed]
- Horan, G.S.; Wood, S.; Ona, V.; Li, D.J.; Lukashev, M.E.; Weinreb, P.H.; Simon, K.J.; Hahm, K.; Allaire, N.E.; Rinaldi, N.J.; et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 2008, 177, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Gai, R.; Chen, Z.; Wang, Y.; Liao, S.; Dong, R.; Zhu, H.; Gu, Y.; He, Q.; Yang, B. The dual PI3K/mTOR inhibitor NVP-BEZ235 prevents epithelial-mesenchymal transition induced by hypoxia and TGF-beta1. Eur. J. Pharmacol. 2014, 729, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Emergence of the phosphoinositide 3-kinase-AKT-mammalian target of rapamycin axis in transforming growth factor-beta-induced epithelial-mesenchymal transition. Cells Tissues Organs 2011, 193, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. Tgf-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Hao, M. Metformin inhibits TGF-beta1-induced epithelial-to-mesenchymal transition via PKM2 relative-mTOR/p70s6k signaling pathway in cervical carcinoma cells. Int. J. Mol. Sci. 2016, 17, 2000. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.Y.; Hao, M. Mammalian target of rapamycin (mTOR) regulates transforming growth factor-beta1 (TGF-beta1)-induced epithelial-mesenchymal transition via decreased pyruvate kinase m2 (pkm2) expression in cervical cancer cells. Med. Sci. Monit. 2017, 23, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Mikaelian, I.; Malek, M.; Gadet, R.; Viallet, J.; Garcia, A.; Girard-Gagnepain, A.; Hesling, C.; Gillet, G.; Gonzalo, P.; Rimokh, R.; et al. Genetic and pharmacologic inhibition of mTORC1 promotes emt by a TGF-beta-independent mechanism. Cancer Res. 2013, 73, 6621–6631. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Autophagy in health and disease. 1. Regulation and significance of autophagy: An overview. Am. J. Physiol. Cell Physiol. 2010, 298, C776–C785. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A.M. Autophagy and disease: Always two sides to a problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.L. Regulation of the autophagy initiating kinase ULK1 by nutrients: Roles of mTORC1 and AMPK. Cell Cycle 2011, 10, 1337–1338. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Ampk and autophagy get connected. EMBO J. 2011, 30, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ulk1 regulates autophagy. PLoS ONE 2010, 5, e15394. [Google Scholar] [CrossRef] [PubMed]
- Romero, Y.; Bueno, M.; Ramirez, R.; Alvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. MTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in ipf fibroblasts. Aging Cell 2016. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Yahalom, J. Pathways that regulate autophagy and their role in mediating tumor response to treatment. Autophagy 2006, 2, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Lee, N.Y.; Nakar, C.; Fitzgerald, M.; Plotkin, J.; Deuel, B.; Hackett, N.; McMahill, M.; Sphicas, E.; Lampen, N.; et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005, 65, 11061–11070. [Google Scholar] [CrossRef] [PubMed]
- Chaachouay, H.; Ohneseit, P.; Toulany, M.; Kehlbach, R.; Multhoff, G.; Rodemann, H.P. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother. Oncol. 2011, 99, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Ondrej, M.; Cechakova, L.; Durisova, K.; Pejchal, J.; Tichy, A. To live or let die: Unclear task of autophagy in the radiosensitization battle. Radiother. Oncol. 2016, 119, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Judge, J.L.; Lacy, S.H.; Ku, W.Y.; Owens, K.M.; Hernady, E.; Thatcher, T.H.; Williams, J.P.; Phipps, R.P.; Sime, P.J.; Kottmann, R.M. The lactate dehydrogenase inhibitor gossypol inhibits radiation-induced pulmonary fibrosis. Radiat. Res. 2017, 188, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Hong, Z.Y.; Nam, J.K.; Lee, H.J.; Jang, J.; Yoo, R.J.; Lee, Y.J.; Lee, C.Y.; Kim, K.H.; Park, S.; et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. 2015, 21, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Westbury, C.B.; Pearson, A.; Nerurkar, A.; Reis-Filho, J.S.; Steele, D.; Peckitt, C.; Sharp, G.; Yarnold, J.R. Hypoxia can be detected in irradiated normal human tissue: A study using the hypoxic marker pimonidazole hydrochloride. Br. J. Radiol. 2007, 80, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Thind, K.; Chen, A.; Friesen-Waldner, L.; Ouriadov, A.; Scholl, T.J.; Fox, M.; Wong, E.; VanDyk, J.; Hope, A.; Santyr, G. Detection of radiation-induced lung injury using hyperpolarized (13)c magnetic resonance spectroscopy and imaging. Magn. Reson. Med. 2013, 70, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.S.; Ouriadov, A.; Thind, K.; Hegarty, E.; Wong, E.; Hope, A.; Santyr, G.E. Detection of radiation induced lung injury in rats using dynamic hyperpolarized (129)Xe magnetic resonance spectroscopy. Med. Phys. 2014, 41, 072302. [Google Scholar] [CrossRef] [PubMed]
- Flockerzi, E.; Schanz, S.; Rube, C.E. Even low doses of radiation lead to DNA damage accumulation in lung tissue according to the genetically-defined DNA repair capacity. Radiother. Oncol. 2014, 111, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra247. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, K.; Araya, J.; Hara, H.; Minagawa, S.; Takasaka, N.; Ito, S.; Kobayashi, K.; Nakayama, K. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir. Investig. 2016, 54, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Ziesche, R.; Fraifeld, V.E. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY) 2015, 7, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Houssaini, A.; Breau, M.; Kebe, K.; Abid, S.; Marcos, E.; Lipskaia, L.; Rideau, D.; Parpaleix, A.; Huang, J.; Amsellem, V.; et al. Mtor pathway activation drives lung cell senescence and emphysema. JCI Insight 2018, 3, 93203. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.G.; Stone, H.B.; Wong, R.S.; Capala, J.; Bernhard, E.J.; Vikram, B.; Coleman, C.N. Normal tissue protection for improving radiotherapy: Where are the gaps? Transl. Cancer Res. 2012, 1, 35–48. [Google Scholar] [PubMed]
- Delanian, S.; Porcher, R.; Rudant, J.; Lefaix, J.L. Kinetics of response to long-term treatment combining pentoxifylline and tocopherol in patients with superficial radiation-induced fibrosis. J. Clin. Oncol. 2005, 23, 8570–8579. [Google Scholar] [CrossRef] [PubMed]
- Haddad, P.; Kalaghchi, B.; Amouzegar-Hashemi, F. Pentoxifylline and vitamin e combination for superficial radiation-induced fibrosis: A phase ii clinical trial. Radiother. Oncol. 2005, 77, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.; Hoglund, P.; Johansson, K.; Jonsson, C.; Killander, F.; Malmstrom, P.; Weddig, A.; Kjellen, E. Pentoxifylline and vitamin e treatment for prevention of radiation-induced side-effects in women with breast cancer: A phase two, double-blind, placebo-controlled randomised clinical trial (ptx-5). Eur. J. Cancer 2009, 45, 2488–2495. [Google Scholar] [CrossRef] [PubMed]
- Anscher, M.S. The irreversibility of radiation-induced fibrosis: Fact or folklore? J. Clin. Oncol. 2005, 23, 8551–8552. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Park, H.J.; Park, Y.S.; Lee, S.M.; Yim, J.J.; Yoo, C.G.; Han, S.K.; Kim, Y.W. Clinical significance of mTOR, ZEB1, ROCK1 expression in lung tissues of pulmonary fibrosis patients. BMC Pulm. Med. 2014, 14, 168. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.S.; Wang, L.; Tian, X.; Li, X.; Ma, A.; Zhou, W.; Zeng, N.; Zhang, J.; Cai, B.; Zhang, H.; et al. Mtor overactivation and compromised autophagy in the pathogenesis of pulmonary fibrosis. PLoS ONE 2015, 10, e0138625. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.X.; Hsieh, A.C.; Kim, W.; Friedlander, T.; Lin, A.M.; Louttit, M.; Ryan, C.J. A phase i study of abiraterone acetate combined with bez235, a dual PI3K/mTOR inhibitor, in metastatic castration resistant prostate cancer. Oncologist 2017, 22, e503–e543. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, M.; Ohzeki, T.; Tamada, S.; Hongo, F.; Anai, S.; Fujimoto, K.; Miki, T.; Nakatani, T.; Fukasawa, S.; Uemura, H. Differences in adverse event profiles between everolimus and temsirolimus and the risk factors for non-infectious pneumonitis in advanced renal cell carcinoma. Int. J. Clin. Oncol. 2015, 20, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, B.J.; Cauley, D.H.; Ng, C.; Millikan, R.E.; Xiao, L.; Corn, P.; Jonasch, E.; Tannir, N.M. Mammalian target of rapamycin (mTOR) inhibitor-associated non-infectious pneumonitis in patients with renal cell cancer: Predictors, management, and outcomes. BJU Int. 2014, 113, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Albiges, L.; Chamming’s, F.; Duclos, B.; Stern, M.; Motzer, R.J.; Ravaud, A.; Camus, P. Incidence and management of mTOR inhibitor-associated pneumonitis in patients with metastatic renal cell carcinoma. Ann. Oncol. 2012, 23, 1943–1953. [Google Scholar] [CrossRef] [PubMed]
- Penttila, P.; Donskov, F.; Rautiola, J.; Peltola, K.; Laukka, M.; Bono, P. Everolimus-induced pneumonitis associates with favourable outcome in patients with metastatic renal cell carcinoma. Eur. J. Cancer 2017, 81, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Popovich, I.G.; Anisimov, V.N.; Zabezhinski, M.A.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Blagosklonny, M.V. Lifespan extension and cancer prevention in her-2/neu transgenic mice treated with low intermittent doses of rapamycin. Cancer Biol. Ther. 2014, 15, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, R.V.; Kondratova, A.A. Rapamycin in preventive (very low) doses. Aging (Albany NY) 2014, 6, 158–159. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Barker, J.; Shi, S.D.; Gehring, M.; Sun, S. Steady-state kinetic and inhibition studies of the mammalian target of rapamycin (mTOR) kinase domain and mTOR complexes. Biochemistry 2010, 49, 8488–8498. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, C.J.; Chen, S.J.; Chin, Y.E. Mtor and post-translational modifications rely on mitochondrion as the arsenal for cellular metabolism regulation. Sci. China Life Sci. 2015, 58, 810–812. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. Mtor: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Mercer, P.F.; Woodcock, H.V.; Eley, J.D.; Plate, M.; Sulikowski, M.G.; Durrenberger, P.F.; Franklin, L.; Nanthakumar, C.B.; Man, Y.; Genovese, F.; et al. Exploration of a potent pi3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in ipf. Thorax 2016, 71, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Corboz, M.R.; Zhang, J.; LaSala, D.; DiPetrillo, K.; Li, Z.; Malinin, V.; Brower, J.; Kuehl, P.J.; Barrett, T.E.; Perkins, W.R.; et al. Therapeutic administration of inhaled ins1009, a treprostinil prodrug formulation, inhibits bleomycin-induced pulmonary fibrosis in rats. Pulm. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.L.; Yao, W.; Guo, Z.J.; Gu, L.; Zhu, Y.J. Low dose pirfenidone suppresses transforming growth factor beta-1 and tissue inhibitor of metalloproteinase-1, and protects rats from lung fibrosis induced by bleomycina. Chin. Med. Sci. J. 2006, 21, 145–151. [Google Scholar] [PubMed]
- Rasooli, R.; Rajaian, H.; Pardakhty, A.; Mandegary, A. Preference of aerosolized pirfenidone to oral intake: An experimental model of pulmonary fibrosis by paraquat. J. Aerosol. Med. Pulm. Drug Deliv. 2018, 31, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Varone, F.; Sgalla, G.; Iovene, B.; Bruni, T.; Richeldi, L. Nintedanib for the treatment of idiopathic pulmonary fibrosis. Expert Opin. Pharmacother. 2018, 19, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Saito, T.; Tanaka, T.; Takoi, H.; Yatagai, Y.; Inomata, M.; Nei, T.; Saito, Y.; Gemma, A.; Azuma, A. Reduced incidence of lung cancer in patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir. Investig. 2018, 56, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Fala, L. Ofev (nintedanib): First tyrosine kinase inhibitor approved for the treatment of patients with idiopathic pulmonary fibrosis. Am. Health Drug Benefits 2015, 8, 101–104. [Google Scholar] [PubMed]
- Kurita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Ichikawa, A.; Saito, N.; Kadota, T.; Tsubouchi, K.; Sato, N.; Yoshida, M.; et al. Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir. Res. 2017, 18, 114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, F.; Kang, L.; Wang, Z.; Wang, Y. Pirfenidone attenuates bleomycin-induced pulmonary fibrosis in mice by regulating nrf2/bach1 equilibrium. BMC Pulm. Med. 2017, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, R.; Ebata, T.; Iwasawa, S.; Ishiwata, T.; Tada, Y.; Tatsumi, K.; Takiguchi, Y. Pirfenidone may revert the epithelial-to-mesenchymal transition in human lung adenocarcinoma. Oncol. Lett. 2017, 14, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, X.; Wang, B.; Nie, Y.; Wen, J.; Wang, Q.; Gu, C. Pirfenidone suppresses mapk signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology (Carlton) 2017, 22, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawrence, J.; Nho, R. The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 778. https://doi.org/10.3390/ijms19030778
Lawrence J, Nho R. The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. International Journal of Molecular Sciences. 2018; 19(3):778. https://doi.org/10.3390/ijms19030778
Chicago/Turabian StyleLawrence, Jessica, and Richard Nho. 2018. "The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis" International Journal of Molecular Sciences 19, no. 3: 778. https://doi.org/10.3390/ijms19030778
APA StyleLawrence, J., & Nho, R. (2018). The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. International Journal of Molecular Sciences, 19(3), 778. https://doi.org/10.3390/ijms19030778