The Modulatory Roles of N-glycans in T-Cell-Mediated Autoimmune Diseases


Abstract
:1. Introduction of Glycosylation
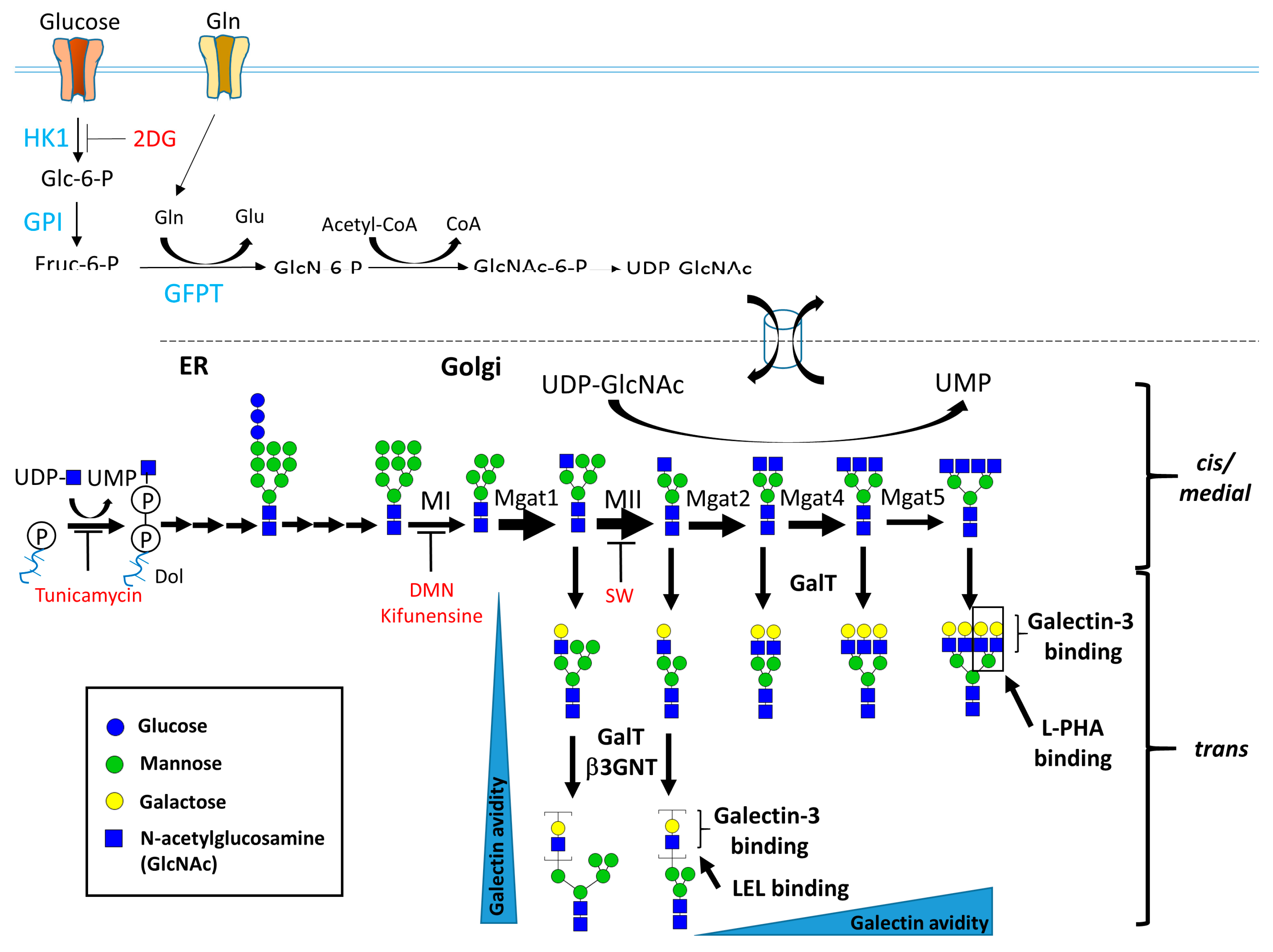
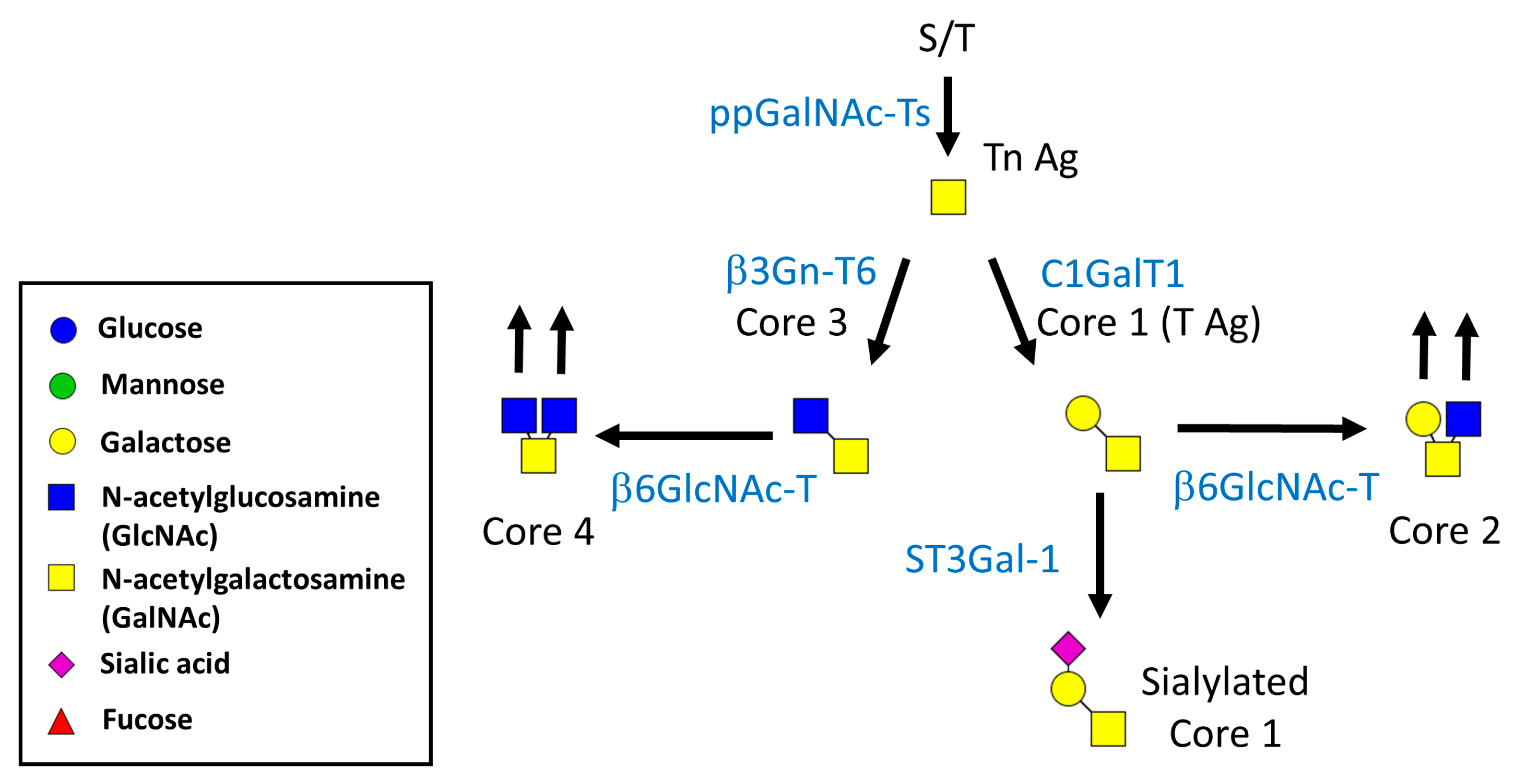
1.1. Biosynthesis of N- and O-Linked Glycosylated Molecules
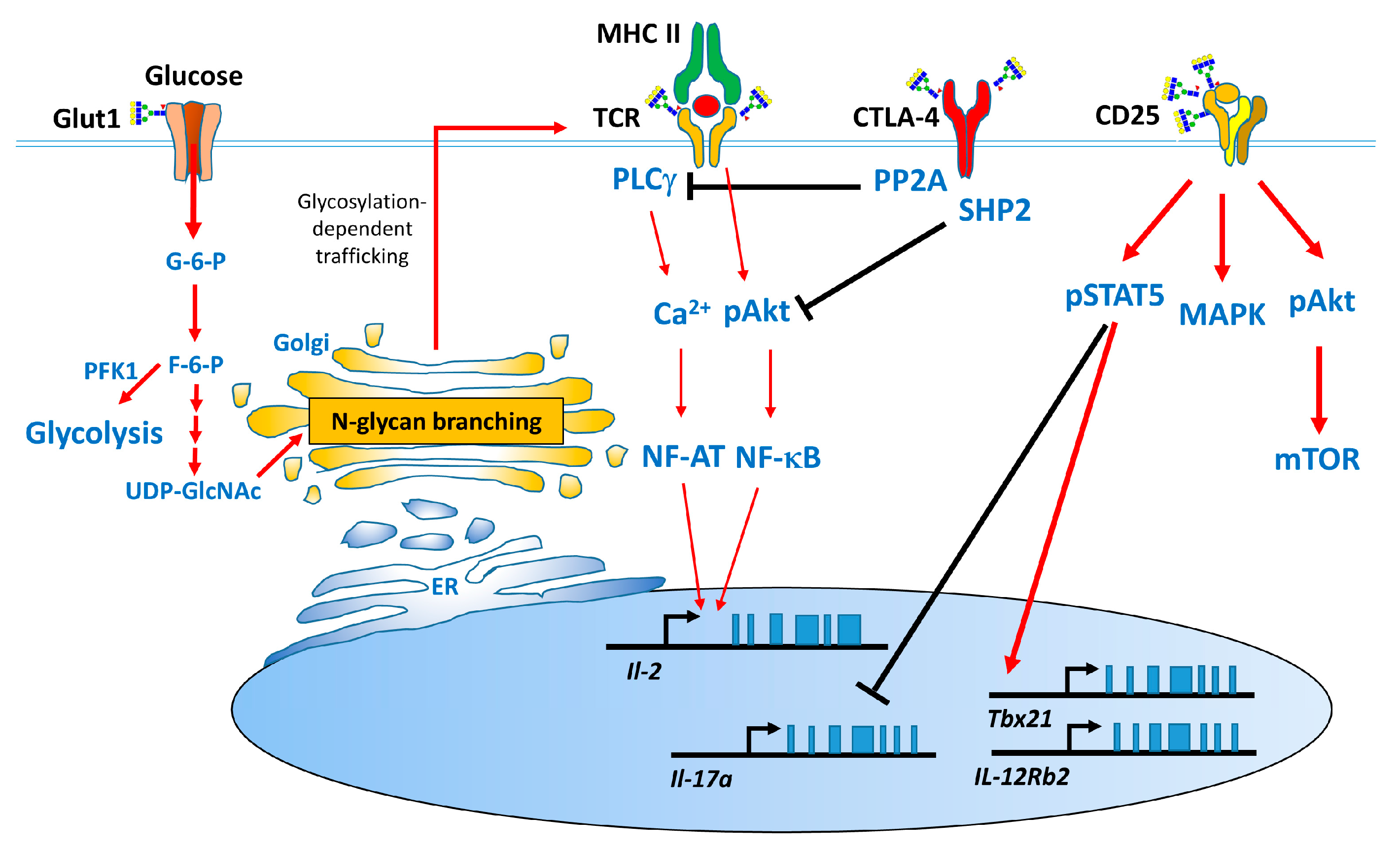
1.2. Biological Functions of N-Glycosylation
1.3. Effect of N-Glycans on the Immune System
2. Overview of Autoimmune Diseases and the Modulatory Effects of N-Glycan Branching on These Diseases
2.1. Multiple Sclerosis
2.1.1. Pathogenesis and Experimental Animal Models of MS
2.1.2. Role of N-Glycan Branching on T Cells in MS
2.2. Systemic Lupus Erythematosus
2.2.1. Pathogenesis and Experimental Animal Models of Systemic Lupus Erythematosus
2.2.2. Role of N-Glycan Branching in SLE
2.3. Inflammatory Bowel Disease
2.3.1. The Pathogenesis and Animal Models for Inflammatory Bowel Disease
2.3.2. Role of Glycosylation on T Cells in IBD
2.4. Type 1 Diabetes Mellitus
2.4.1. Pathogenesis of Type 1 Diabetes Mellitus and Non-Obese Diabetic Mice
2.4.2. Role of N-Glycans in T1D
3. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| EAE | Experimental autoimmune encephalomyelitis |
| HBP | Hexosamine biosynthesis pathway |
| SLE | Systemic lupus erythematosus |
| IBD | Inflammatory bowel disease |
| T1D | Type 1 diabetes mellitus |
| NOD | Non-obese diabetic |
| DSS | Dextran sodium sulfate |
| TNBS | Trinitrobenzene sulfonic acid |
References
- Sasai, K.; Ikeda, Y.; Fujii, T.; Tsuda, T.; Taniguchi, N. UDP-GlcNAc concentration is an important factor in the biosynthesis of β1,6-branched oligosaccharides: Regulation based on the kinetic properties of N-acetylglucosaminyltransferase V. Glycobiology 2002, 12, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Mkhikian, H.; Grigorian, A.; Li, C.F.; Chen, H.L.; Newton, B.; Zhou, R.W.; Beeton, C.; Torossian, S.; Tatarian, G.G.; Lee, S.U.; et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat. Commun. 2011, 2, 334. [Google Scholar] [CrossRef] [PubMed]
- Boscher, C.; Dennis, J.W.; Nabi, I.R. Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 2011, 23, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.D.; Liu, F.T. Galectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor: New York, NY, USA, 2009. [Google Scholar]
- Schnaar, R.L. Glycobiology simplified: Diverse roles of glycan recognition in inflammation. J. Leukoc. Biol. 2016, 99, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, J.; Bao, X.; Petryanik, B.; Schaerli, P.; Gauguet, J.M.; Yu, S.Y.; Kawashima, H.; Saito, H.; Ohtsubo, K.; Marth, J.D.; et al. Critical functions of N-glycans in l-selectin-mediated lymphocyte homing and recruitment. Nat. Immunol. 2007, 8, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.D. Ligands for l-selectin: Homing, inflammation, and beyond. Annu. Rev. Immunol. 2004, 22, 129–156. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef] [PubMed]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.T.; Ten Hagen, K.G. Mucin-type O-glycosylation during development. J. Biol. Chem. 2013, 288, 6921–6929. [Google Scholar] [CrossRef] [PubMed]
- Ellies, L.G.; Tsuboi, S.; Petryniak, B.; Lowe, J.B.; Fukuda, M.; Marth, J.D. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity 1998, 9, 881–890. [Google Scholar] [CrossRef]
- Homeister, J.W.; Thall, A.D.; Petryniak, B.; Maly, P.; Rogers, C.E.; Smith, P.L.; Kelly, R.J.; Gersten, K.M.; Askari, S.W.; Cheng, G.; et al. The α(1,3)fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity 2001, 15, 115–126. [Google Scholar] [CrossRef]
- Ju, T.; Brewer, K.; D’Souza, A.; Cummings, R.D.; Canfield, W.M. Cloning and expression of human core 1 β1,3-galactosyltransferase. J. Biol. Chem. 2002, 277, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.; Cummings, R.D.; Canfield, W.M. Purification, characterization, and subunit structure of rat core 1 β1,3-galactosyltransferase. J. Biol. Chem. 2002, 277, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.; Cummings, R.D. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 β3-galactosyltransferase. Proc. Natl. Acad. Sci. USA 2002, 99, 16613–16618. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, Y.; Ikehara, Y.; Iwasaki, H.; Nonomura, C.; Sato, T.; Nakanishi, H.; Narimatsu, H. Immunocytochemical analysis for intracellular dynamics of C1GalT associated with molecular chaperone, Cosmc. Biochem. Biophys. Res. Commun. 2008, 366, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ju, T.; Ding, X.; Xia, B.; Wang, W.; Xia, L.; He, M.; Cummings, R.D. Cosmc is an essential chaperone for correct protein O-glycosylation. Proc. Natl. Acad. Sci. USA 2010, 107, 9228–9233. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.K.; Yang, R.Y.; Saegusa, J.; Liu, F.T. Analysis of the intracellular role of galectins in cell growth and apoptosis. Methods Mol. Biol. 2015, 1207, 451–463. [Google Scholar] [PubMed]
- Yang, R.Y.; Rabinovich, G.A.; Liu, F.T. Galectins: Structure, function and therapeutic potential. Expert Rev. Mol. Med. 2008, 10, e17. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Dumic, J.; Dabelic, S.; Flogel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.T.; Lee, Y.C. Affinity enhancement by multivalent lectin-carbohydrate interaction. Glycoconj. J. 2000, 17, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.F.; Miceli, M.C.; Baum, L.G. Clusters, bundles, arrays and lattices: Novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr. Opin. Struct. Biol. 2002, 12, 616–623. [Google Scholar] [CrossRef]
- Ahmad, N.; Gabius, H.J.; Andre, S.; Kaltner, H.; Sabesan, S.; Roy, R.; Liu, B.; Macaluso, F.; Brewer, C.F. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J. Biol. Chem. 2004, 279, 10841–10847. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, M.; Granovsky, M.; Quaggin, S.; Dennis, J.W. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 2001, 409, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Mkhikian, H.; Mortales, C.L.; Zhou, R.W.; Khachikyan, K.; Wu, G.; Haslam, S.M.; Kavarian, P.; Dell, A.; Demetriou, M. Golgi self-correction generates bioequivalent glycans to preserve cellular homeostasis. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Ioffe, E.; Stanley, P. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc. Natl. Acad. Sci. USA 1994, 91, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Metzler, M.; Gertz, A.; Sarkar, M.; Schachter, H.; Schrader, J.W.; Marth, J.D. Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J. 1994, 13, 2056–2065. [Google Scholar] [PubMed]
- Wang, Y.; Tan, J.; Sutton-Smith, M.; Ditto, D.; Panico, M.; Campbell, R.M.; Varki, N.M.; Long, J.M.; Jaeken, J.; Levinson, S.R.; et al. Modeling human congenital disorder of glycosylation type IIa in the mouse: Conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology 2001, 11, 1051–1070. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Dunn, J.; Jaeken, J.; Schachter, H. Mutations in the MGAT2 gene controlling complex N-glycan synthesis cause carbohydrate-deficient glycoprotein syndrome type II, an autosomal recessive disease with defective brain development. Am. J. Hum. Genet. 1996, 59, 810–817. [Google Scholar] [PubMed]
- Ohtsubo, K.; Takamatsu, S.; Minowa, M.T.; Yoshida, A.; Takeuchi, M.; Marth, J.D. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 2005, 123, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, K.; Chen, M.Z.; Olefsky, J.M.; Marth, J.D. Pathway to diabetes through attenuation of pancreatic β cell glycosylation and glucose transport. Nat. Med. 2011, 17, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, M.; Fata, J.; Pawling, J.; Muller, W.J.; Khokha, R.; Dennis, J.W. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat. Med. 2000, 6, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Pawling, J.; Partridge, E.A.; Sukhu, B.; Grynpas, M.; Dennis, J.W. Metabolic homeostasis and tissue renewal are dependent on β1,6GlcNAc-branched N-glycans. Glycobiology 2007, 17, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Green, R.S.; Stone, E.L.; Tenno, M.; Lehtonen, E.; Farquhar, M.G.; Marth, J.D. Mammalian N-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity 2007, 27, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.W.; Mkhikian, H.; Grigorian, A.; Hong, A.; Chen, D.; Arakelyan, A.; Demetriou, M. N-glycosylation bidirectionally extends the boundaries of thymocyte positive selection by decoupling Lck from Ca2+ signaling. Nat. Immunol. 2014, 15, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Gao, G.; Pawling, J.; Dennis, J.W.; Demetriou, M.; Li, B. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J. Immunol. 2004, 173, 7200–7208. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, A.; Araujo, L.; Naidu, N.N.; Place, D.J.; Choudhury, B.; Demetriou, M. N-acetylglucosamine inhibits T-helper 1 (Th1)/T-helper 17 (Th17) cell responses and treats experimental autoimmune encephalomyelitis. J. Biol. Chem. 2011, 286, 40133–40141. [Google Scholar] [CrossRef] [PubMed]
- Anjos, S.; Nguyen, A.; Ounissi-Benkalha, H.; Tessier, M.C.; Polychronakos, C. A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. J. Biol. Chem. 2002, 277, 46478–46486. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Loserth, S.; Kolb-Maurer, A.; Ponath, A.; Wiese, S.; Kruse, N.; Rieckmann, P. A polymorphism in the human cytotoxic T-lymphocyte antigen 4 (CTLA4) gene (exon 1 +49) alters T-cell activation. Immunogenetics 2002, 54, 1–8. [Google Scholar] [PubMed]
- Minami, Y.; Kono, T.; Miyazaki, T.; Taniguchi, T. The IL-2 receptor complex: Its structure, function, and target genes. Annu. Rev. Immunol. 1993, 11, 245–268. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Paul, W.E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.W.; Lin, M.H.; Huang, S.H.; Fu, S.H.; Hsu, C.Y.; Yen, B.L.; Chen, J.T.; Chang, D.M.; Sytwu, H.K. Glucosamine Modulates T Cell Differentiation through Down-regulating N-Linked Glycosylation of CD25. J. Biol. Chem. 2015, 290, 29329–29344. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.; Khim, P.; Mkhikian, H.; Mortales, C.L.; Demetriou, M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.; Diamond, B. Autoimmune diseases. N. Engl. J. Med. 2001, 345, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 85, 299–302. [Google Scholar] [CrossRef]
- Sobel, R.A.; Blanchette, B.W.; Bhan, A.K.; Colvin, R.B. The immunopathology of experimental allergic encephalomyelitis. II. Endothelial cell Ia increases prior to inflammatory cell infiltration. J. Immunol. 1984, 132, 2402–2407. [Google Scholar] [PubMed]
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, A.M.; Miller, S.D. Mechanisms of immunopathology in murine models of central nervous system demyelinating disease. J. Immunol. 2006, 176, 3293–3298. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.; Karpus, W.J. Experimental autoimmune encephalomyelitis in the mouse. Curr. Protoc. Immunol. 2007. [Google Scholar] [CrossRef]
- Kennedy, K.J.; Karpus, W.J. Role of chemokines in the regulation of Th1/Th2 and autoimmune encephalomyelitis. J. Clin. Immunol. 1999, 19, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T. Cytokines in neuroinflammatory disease: Role of myelin autoreactive T cell production of interferon-γ. J. Neuroimmunol. 1992, 40, 211–218. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Grigorian, A.; Pawling, J.; Chen, I.J.; Gao, G.; Mozaffar, T.; McKerlie, C.; Demetriou, M. N-glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. J. Biol. Chem. 2007, 282, 33725–33734. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ashe, S.; Brady, W.A.; Hellstrom, I.; Hellstrom, K.E.; Ledbetter, J.A.; McGowan, P.; Linsley, P.S. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell 1992, 71, 1093–1102. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Alegre, M.L.; Frauwirth, K.A.; Thompson, C.B. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001, 1, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Suh, H.N.; Kim, M.O.; Ryu, J.M.; Han, H.J. Glucosamine-induced OGT activation mediates glucose production through cleaved Notch1 and FoxO1, which coordinately contributed to the regulation of maintenance of self-renewal in mouse embryonic stem cells. Stem Cells Dev. 2014, 23, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Macauley, M.S.; Vocadlo, D.J. Increasing O-GlcNAc levels: An overview of small-molecule inhibitors of O-GlcNAcase. Biochim. Biophys. Acta 2010, 1800, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Liang, C.M.; Chen, Y.H.; Tai, M.C.; Lu, D.W.; Chen, J.T. Glucosamine modulates TNF-α-induced ICAM-1 expression and function through O-linked and N-linked glycosylation in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Klenk, H.D.; Scholtissek, C.; Rott, R. Inhibition of glycoprotein biosynthesis of influenza virus by d-glucosamine and 2-deoxy-d-glucose. Virology 1972, 49, 723–734. [Google Scholar] [CrossRef]
- Forchhammer, L.; Thorn, M.; Met, O.; Gad, M.; Weidner, M.S.; Claesson, M.H. Immunobiological effects of glucosamine in vitro. Scand. J. Immunol. 2003, 58, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Cheong, K.A.; Park, C.D.; Lee, A.Y. Glucosamine improved atopic dermatitis-like skin lesions in NC/Nga mice by inhibition of Th2 cell development. Scand. J. Immunol. 2011, 73, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Rudert, W.A.; Harnaha, J.; Wright, M.; Machen, J.; Lakomy, R.; Qian, S.; Lu, L.; Robbins, P.D.; Trucco, M.; et al. Immunosuppressive effects of glucosamine. J. Biol. Chem. 2002, 277, 39343–39349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.X.; Yu, S.; Gran, B.; Rostami, A. Glucosamine abrogates the acute phase of experimental autoimmune encephalomyelitis by induction of Th2 response. J. Immunol. 2005, 175, 7202–7208. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.; Lai, K.N. Cytokines and their roles in the pathogenesis of systemic lupus erythematosus: From basics to recent advances. J. Biomed. Biotechnol. 2010, 2010, 365083. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T. Therapeutic strategies for SLE involving cytokines: Mechanism-oriented therapies especially IFN-γ targeting gene therapy. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Lit, L.C.; Tam, L.S.; Li, E.K.; Wong, P.T.; Lam, C.W. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: Implications for Th17-mediated inflammation in auto-immunity. Clin. Immunol. 2008, 127, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Dixon, F.J. Murine models of systemic lupus erythematosus. Adv. Immunol. 1985, 37, 269–390. [Google Scholar] [PubMed]
- Watson, M.L.; Rao, J.K.; Gilkeson, G.S.; Ruiz, P.; Eicher, E.M.; Pisetsky, D.S.; Matsuzawa, A.; Rochelle, J.M.; Seldin, M.F. Genetic analysis of MRL-lpr mice: Relationship of the Fas apoptosis gene to disease manifestations and renal disease-modifying loci. J. Exp. Med. 1992, 176, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.; Sang, A.; Yin, Y.; Zheng, Y.Y.; Morel, L. Murine models of systemic lupus erythematosus. J. Biomed. Biotechnol. 2011, 2011, 271694. [Google Scholar] [CrossRef] [PubMed]
- Chui, D.; Sellakumar, G.; Green, R.; Sutton-Smith, M.; McQuistan, T.; Marek, K.; Morris, H.; Dell, A.; Marth, J. Genetic remodeling of protein glycosylation in vivo induces autoimmune disease. Proc. Natl. Acad. Sci. USA 2001, 98, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Mizoguchi, E. Animal models of IBD: Linkage to human disease. Curr. Opin. Pharmacol. 2010, 10, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Nagahama, K.; Imaeda, H.; Ogawa, A.; Lau, C.W.; Kobayashi, T.; Hisamatsu, T.; Preffer, F.I.; Mizoguchi, E.; Ikeuchi, H.; et al. Inducible colitis-associated glycome capable of stimulating the proliferation of memory CD4+ T cells. J. Exp. Med. 2012, 209, 2383–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, A.M.; Dourado, J.; Lago, P.; Cabral, J.; Marcos-Pinto, R.; Salgueiro, P.; Almeida, C.R.; Carvalho, S.; Fonseca, S.; Lima, M.; et al. Dysregulation of T cell receptor N-glycosylation: A molecular mechanism involved in ulcerative colitis. Hum. Mol. Genet. 2014, 23, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Shinzaki, S.; Iijima, H.; Wakamatsu, K.; Iwamoto, C.; Sobajima, T.; Kuwahara, R.; Hiyama, S.; Hayashi, Y.; Takamatsu, S.; et al. Core Fucosylation on T Cells, Required for Activation of T-Cell Receptor Signaling and Induction of Colitis in Mice, Is Increased in Patients With Inflammatory Bowel Disease. Gastroenterology 2016, 150, 1620–1632. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, E.; Abiru, N.; Eguchi, K. Prevention of type 1 diabetes: From the view point of β cell damage. Diabetes Res. Clin. Pract. 2004, 66 (Suppl. S1), S27–S32. [Google Scholar] [CrossRef] [PubMed]
- Wicker, L.S.; Todd, J.A.; Peterson, L.B. Genetic control of autoimmune diabetes in the NOD mouse. Annu. Rev. Immunol. 1995, 13, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Furio, L.; Mecheri, R.; van der Does, A.M.; Lundeberg, E.; Saveanu, L.; Chen, Y.; van Endert, P.; Agerberth, B.; Diana, J. Pancreatic β-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed under the Influence of the Gut Microbiota. Immunity 2015, 43, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C.A.; Borchers, A.T.; Ridgway, W.M.; Keen, C.L.; Ansari, A.A.; Gershwin, M.E. NOD mice and autoimmunity. Autoimmun. Rev. 2005, 4, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Fox, H.S. Androgen treatment prevents diabetes in nonobese diabetic mice. J. Exp. Med. 1992, 175, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Debray-Sachs, M.; Carnaud, C.; Boitard, C.; Cohen, H.; Gresser, I.; Bedossa, P.; Bach, J.F. Prevention of diabetes in NOD mice treated with antibody to murine IFN γ. J. Autoimmun. 1991, 4, 237–248. [Google Scholar] [CrossRef]
- Yi, Z.; Li, L.; Garland, A.; He, Q.; Wang, H.; Katz, J.D.; Tisch, R.; Wang, B. IFN-γ receptor deficiency prevents diabetes induction by diabetogenic CD4+, but not CD8+, T cells. Eur. J. Immunol. 2012, 42, 2010–2018. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.J.; Arreaza, G.A.; Zucker, P.; Chensue, S.W.; Strieter, R.M.; Chakrabarti, S.; Delovitch, T.L. IL-4 prevents insulitis and insulin-dependent diabetes mellitus in nonobese diabetic mice by potentiation of regulatory T helper-2 cell function. J. Immunol. 1997, 159, 4686–4692. [Google Scholar] [PubMed]
- Pennline, K.J.; Roque-Gaffney, E.; Monahan, M. Recombinant human IL-10 prevents the onset of diabetes in the nonobese diabetic mouse. Clin. Immunol. Immunopathol. 1994, 71, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, A.; Lee, S.U.; Tian, W.; Chen, I.J.; Gao, G.; Mendelsohn, R.; Dennis, J.W.; Demetriou, M. Control of T Cell-mediated autoimmunity by metabolite flux to N-glycan biosynthesis. J. Biol. Chem. 2007, 282, 20027–20035. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, C.F.; Mkhikian, H.; Zhou, R.W.; Newton, B.L.; Demetriou, M. Family studies of type 1 diabetes reveal additive and epistatic effects between MGAT1 and three other polymorphisms. Genes Immun. 2014, 15, 218–223. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease | Strategies Used | Target Glycans | Clinical Outcome | Mechanisms | Ref. |
|---|---|---|---|---|---|
| EAE | T-cell specific Mgat2 KO mice | Defective N-glycan branching | Increase severity | Increased TCR clustering and CTLA-4 endocytosis | [27] |
 | |||||
| Mgat5 KO mice | Defective N-glycan branching | Increase severity | Increased TCR clustering and CTLA-4 endocytosis | [2,26] | |
 | |||||
| Administration of vitamin D | Enhance N-glycan branching | Reduced severity | Increased CTLA-4 surface retention | [3] | |
 | |||||
| Administration of GlcNAc | Enhance N-glycan branching | Reduced severity | Decreased Th1 and Th17 cell responses | [40] | |
 | |||||
| Administration of GlcN | Inhibition of N-glycosylation | Increase severity | Increased Th17 response via the decrease of CD25 surface retention | [45] | |
| MS | N-glycan branching | Risk factor | Increased the TCR clustering and decreased CTLA-4 surface retention (Mgat1 haplotype) | [3] | |
 | |||||
| Decrease N-glycans site of CTLA-4 | Risk factor | Increased CTLA-4 endocytosis (CTLA-4 SNP) | [3] | ||
| SLE | α-mannosidase II KO mice | N-glycan branching | Increase severity | Increased innate immunity | [37,75] |
 | |||||
| Mgat5 KO mice | N-glycan branching | Increase severity | Unknown | [26] | |
 | |||||
| IBD | Fut8 KO mice with DSS, TNBS and cell transfer-induced colitis | Defective Core fucosylation | Reduced severity | Decreased TCR signaling | [82] |
 | |||||
| T-cell specific Tg C2GnT mice | Defective C2GnT | Reduced severity | Increased Immunological synapses | [80] | |
 | |||||
| T1D | Administration of GlcNAc | N-glycan branching | Reduced severity | Decreased Th1 responses | [92] |
 | |||||
| Administration of GlcN | Inhibition of N-glycosylation | Reduced severity | Decreased Th1 response via the downregulation of CD25 and Glut1 surface retention | [45] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chien, M.-W.; Fu, S.-H.; Hsu, C.-Y.; Liu, Y.-W.; Sytwu, H.-K. The Modulatory Roles of N-glycans in T-Cell-Mediated Autoimmune Diseases. Int. J. Mol. Sci. 2018, 19, 780. https://doi.org/10.3390/ijms19030780
Chien M-W, Fu S-H, Hsu C-Y, Liu Y-W, Sytwu H-K. The Modulatory Roles of N-glycans in T-Cell-Mediated Autoimmune Diseases. International Journal of Molecular Sciences. 2018; 19(3):780. https://doi.org/10.3390/ijms19030780
Chicago/Turabian StyleChien, Ming-Wei, Shin-Huei Fu, Chao-Yuan Hsu, Yu-Wen Liu, and Huey-Kang Sytwu. 2018. "The Modulatory Roles of N-glycans in T-Cell-Mediated Autoimmune Diseases" International Journal of Molecular Sciences 19, no. 3: 780. https://doi.org/10.3390/ijms19030780
APA StyleChien, M. -W., Fu, S. -H., Hsu, C. -Y., Liu, Y. -W., & Sytwu, H. -K. (2018). The Modulatory Roles of N-glycans in T-Cell-Mediated Autoimmune Diseases. International Journal of Molecular Sciences, 19(3), 780. https://doi.org/10.3390/ijms19030780