Understanding Splenomegaly in Myelofibrosis: Association with Molecular Pathogenesis

Abstract
:1. Introduction
2. Somatic Mutations Associated with the Pathogenesis of MF
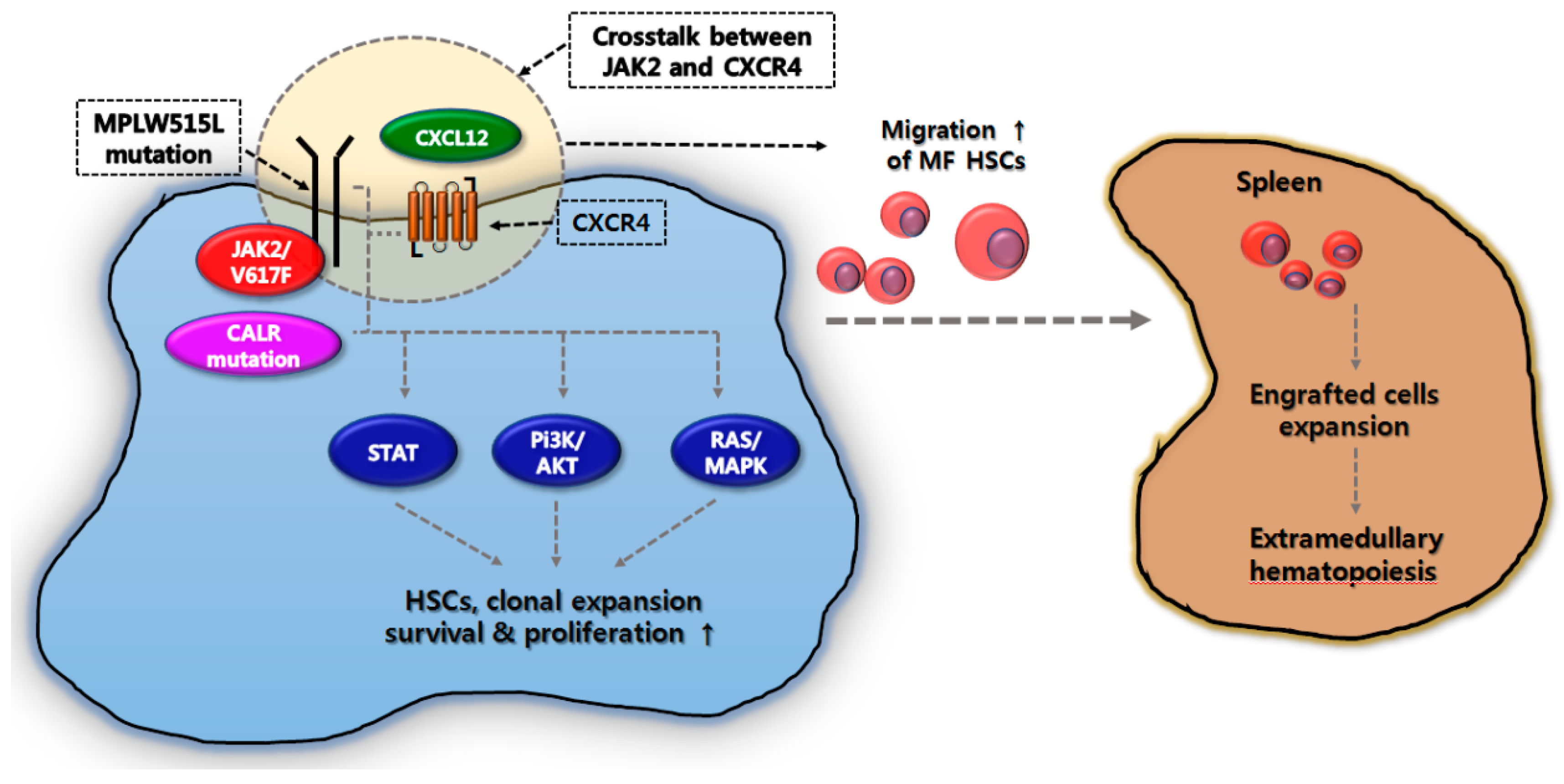
3. Correlation between EMH in the Spleen and Molecular Pathogenesis in MF
4. Gene Mutations Associated with Splenomegaly in MF
5. Reduction in Splenomegaly as a Benefit of JAK Inhibitors in MF
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Tefferi, A.; Thiele, J.; Orazi, A.; Kvasnicka, H.M.; Barbui, T.; Hanson, C.A.; Barosi, G.; Verstovsek, S.; Birgegard, G.; Mesa, R.; et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: Recommendations from an ad hoc international expert panel. Blood 2007, 110, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, R.J.; Salo, A.; Fialkow, P.J. Agnogenic myeloid metaplasia: A clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood 1978, 51, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G. Myelofibrosis with myeloid metaplasia: Diagnostic definition and prognostic classification for clinical studies and treatment guidelines. J. Clin. Oncol. 1999, 17, 2954–2970. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Nagorney, D.S.; Schwager, S.; Allred, J.; Tefferi, A. Palliative goals, patient selection, and perioperative platelet management: Outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer 2006, 107, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.D.; Thompson, C.A.; Wang, A.H.; Vierkant, R.A.; Habermann, T.M.; Ross, J.A.; Mesa, R.A.; Virnig, B.A.; Cerhan, J.R. Anthropometric, medical history and lifestyle risk factors for myeloproliferative neoplasms in the Iowa Women’s Health Study cohort. Int. J. Cancer 2014, 134, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Savastano, S.; Di Somma, C.; Pizza, G.; De Rosa, A.; Nedi, V.; Rossi, A.; Orio, F.; Lombardi, G.; Colao, A.; Tarantino, G. Liver-spleen axis, insulin-like growth factor-(IGF)-I axis and fat mass in overweight/obese females. J. Transl. Med. 2011, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C. Genetic and epigenetic complexity in myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Guerini, V.; Barosi, G.; Ruggeri, M.; Specchia, G.; Lo-Coco, F.; Delaini, F.; et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood 2008, 112, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SRSF2 mutations in primary myelofibrosis: Significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Skoda, R.C. Molecular pathogenesis of Philadelphia chromosome negative myeloproliferative disorders. Blood Rev. 2005, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Inra, C.N.; Zhou, B.O.; Acar, M.; Murphy, M.M.; Richardson, J.; Zhao, Z.; Morrison, S.J. A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature 2015, 527, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Le Bousse-Kerdilès, M.C.; Martyré, M.C. Dual implication of fibrogenic cytokines in the pathogenesis of fibrosis and myeloproliferation in myeloid metaplasia with myelofibrosis. Ann. Hematol. 1999, 78, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, J.J.; Pierre-Louis, O.; Hasselbalch, H.C.; Uzan, G.; Jasmin, C.; Martyré, M.C.; Le Bousse-Kerdilès, M.C. French INSERM and the European EUMNET Networks on Myelofibrosis. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood 2008, 112, 3026–3035. [Google Scholar] [CrossRef] [PubMed]
- Emadi, S.; Clay, D.; Desterke, C.; Guerton, B.; Maquarre, E.; Charpentier, A.; Jasmin, C.; Le Bousse-Kerdilès, M.C. French INSERM Research Network on MMM. IL-8 and its CXCR1 and CXCR2 receptors participate in the control of megakaryocytic proliferation, differentiation, and ploidy in myeloid metaplasia with myelofibrosis. Blood 2005, 105, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Petit, I. Current understanding of stem cell mobilization: The roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp. Hematol. 2002, 30, 973–981. [Google Scholar] [CrossRef]
- Petit, I.; Szyper-Kravitz, M.; Nagler, A.; Lahav, M.; Peled, A.; Habler, L.; Ponomaryov, T.; Taichman, R.S.; Arenzana-Seisdedos, F.; Fujii, N.; et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat. Immunol. 2002, 3, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Miwa, Y.; Hayashi, T.; Suzuki, S.; Abe, S.; Onishi, I.; Kirimura, S.; Kitagawa, M.; Kurata, M. Up-regulated expression of CXCL12 in human spleens with extramedullary haematopoiesis. Pathology 2013, 45, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Xu, M.; Roboz, J.; Lu, M.; Mascarenhas, J.; Hoffman, R. The effect of CXCL12 processing on CD34+ cell migration in myeloproliferative neoplasms. Cancer Res. 2010, 70, 3402–3410. [Google Scholar] [CrossRef] [PubMed]
- Bogani, C.; Ponziani, V.; Guglielmelli, P.; Desterke, C.; Rosti, V.; Bosi, A.; Le Bousse-Kerdilès, M.C.; Barosi, G.; Vannucchi, A.M. Myeloproliferative Disorders Research Consortium. Hypermethylation of CXCR4 promoter in CD34+ cells from patients with primary myelofibrosis. Stem Cells 2008, 26, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Rosti, V.; Massa, M.; Vannucchi, A.M.; Bergamaschi, G.; Campanelli, R.; Pecci, A.; Viarengo, G.; Meli, V.; Marchetti, M.; Guglielmelli, P.; et al. Italian Registry of Myelofibrosis with Myeloid Metaplasia; Myeloproliferative Disorders Research Consortium. The expression of CXCR4 is down-regulated on the CD34+ cells of patients with myelofibrosis with myeloid metaplasia. Blood. Cells Mol. Dis. 2007, 38, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.R.; Martelli, F.; Verrucci, M.; Migliaccio, G.; Vannucchi, A.M.; Ni, H.; Xu, M.; Jiang, Y.; Nakamoto, B.; Papayannopoulou, T.; et al. Altered SDF-1/CXCR4 axis in patients with primary myelofibrosis and in the Gata1 low mouse model of the disease. Exp. Hematol. 2008, 360, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Wang, J.F.; Matczak, E.; Proper, J.A.; Groopman, J.E. Janus kinase 2 is involved in stromal cell-derived factor-1alpha-induced tyrosine phosphorylation of focal adhesion proteins and migration of hematopoietic progenitor cells. Blood 2001, 97, 3342–3348. [Google Scholar] [CrossRef] [PubMed]
- Abdelouahab, H.; Zhang, Y.; Wittner, M.; Oishi, S.; Fujii, N.; Besancenot, R.; Plo, I.; Ribrag, V.; Solary, E.; Vainchenker, W.; et al. CXCL12/CXCR4 pathway is activated by oncogenic JAK2 in a PI3K-dependent manner. Oncotarget 2016, 8, 54082–54095. [Google Scholar] [CrossRef] [PubMed]
- Cantor, A.B.; Orkin, S.H. Transcriptional regulation of erythropoiesis: An affair involving multiple partners. Oncogene 2002, 21, 3368–3376. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Pancrazzi, A.; Guglielmelli, P.; Di Lollo, S.; Bogani, C.; Baroni, G.; Bianchi, L.; Migliaccio, A.R.; Bosi, A.; Paoletti, F. Abnormalities of GATA-1 in megakaryocytes from patients with idiopathic myelofibrosis. Am. J. Pathol. 2005, 167, 849–858. [Google Scholar] [CrossRef]
- Zingariello, M.; Sancillo, L.; Martelli, F.; Ciaffoni, F.; Marra, M.; Varricchio, L.; Rana, R.A.; Zhao, C.; Crispino, J.D.; Migliaccio, A.R. The thrombopoietin/MPL axis is activated in the Gata1low mouse model of myelofibrosis and is associated with a defective RPS14 signature. Blood Cancer J. 2017, 7, e572. [Google Scholar] [CrossRef] [PubMed]
- Gilles, L.; Arslan, A.D.; Marinaccio, C.; Wen, Q.J.; Arya, P.; McNulty, M.; Yang, Q.; Zhao, J.C.; Konstantinoff, K.; Lasho, T.; et al. Downregulation of GATA1 drives impaired hematopoiesis in primary myelofibrosis. J. Clin. Investig. 2017, 127, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Verrucci, M.; Pancrazzi, A.; Aracil, M.; Martelli, F.; Guglielmelli, P.; Zingariello, M.; Ghinassi, B.; D’Amore, E.; Jimeno, J.; Vannucchi, A.M.; et al. CXCR4-independent rescue of the myeloproliferative defect of the Gata1low myelofibrosis mouse model by Aplidin. J. Cell. Physiol. 2010, 225, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Bergamaschi, G.; Marchetti, M.; Vannucchi, A.M.; Guglielmelli, P.; Antonioli, E.; Massa, M.; Rosti, V.; Campanelli, R.; Villani, L.; et al. Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto (GIMEMA) Italian Registry of Myelofibrosis. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood 2007, 110, 4030–4036. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martínez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; Newberry, K.J.; Luthra, R.; Jabbour, E.; Pierce, S.; Cortes, J.; Singh, R.; Mehrotra, M.; Routbort, M.J.; Luthra, M.; et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015, 126, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; Seymour, J.F.; Roberts, A.W.; Wadleigh, M.; To, L.B.; Scherber, R.; Turba, E.; Dorr, A.; Zhu, J.; Wang, L.; et al. Results of a phase 2 study of pacritinib (SB1518), a JAK2/JAK2(V617F) inhibitor, in patients with myelofibrosis. Blood 2015, 125, 2649–2655. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Vannucchi, A.M.; Mead, A.; Egyed, M.; Szoke, A.; Suvorov, A.; Jakucs, J.; Perkins, A.; Prasad, R.; Mayer, J.; et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): An international, randomised, phase 3 trial. Lancet Haematol. 2017, 4, e225–e236. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Aaron, G.T.; Stein, B.; Gupta Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Results of the Persist-2 Phase 3 Study of Pacritinib (PAC) Versus Best Available Therapy (BAT), Including Ruxolitinib (RUX), in Patients (pts) with Myelofibrosis (MF) and Platelet Counts <100,000/µL. Blood 2016, 128, LBA-5. [Google Scholar]
- Mesa, R.A.; Kiladjian, J.J.; Catalano, J.V.; Devos, T.; Egyed, M.; Hellman, A. Phase 3 trial of momelotinib (MMB) vs. ruxolitinib (RUX) in JAK inhibitor (JAKi) naive patients with myelofibrosis (MF). J. Clin. Oncol. 2017, 35, 7000. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D. Phase 3 randomized trial of momelotinib (MMB) versus best available therapy (BAT) in patients with myelofibrosis (MF) previously treated with ruxolitinib (RUX). J. Clin. Oncol. 2017, 35, 7001. [Google Scholar]
- Zhou, T.; Georgeon, S.; Moser, R.; Moore, D.J.; Caflisch, A.; Hantschel, O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia 2014, 28, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Harrison, C.N.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Results of a randomized, double-blind, placebo-controlled phase III study (JAKARTA) of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis (MF). Blood 2013, 122, 393. [Google Scholar]
- Tefferi, A. Primary myelofibrosis: 2013 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2013, 88, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Mudireddy, M.; Gangat, N.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Nagorney, D.M. Risk factors and a prognostic model for postsplenectomy survival in myelofibrosis. Am. J. Hematol. 2017, 92, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (international working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS plus: A refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Schwager, S.; Huang, J.; Pardanani, A.D.; Hussein, K.; Camoriano, J.; Tefferi, A. Weight Loss, Splenomegaly, and Hypocholesterolemia in Myeloproliferative Neoplasms: Patterns and Relevance from the Pre JAK2 Inhibitor Era. Blood 2009, 114, 3918. [Google Scholar]
- Wang, J.; Xu, J.; Gale, R.P.; Xu, Z.; Li, B.; Qin, T.; Zhang, Y.; Fang, L.; Zhang, H.; Pan, L.; et al. Prognostic impact of splenomegaly on survival of Chinese with primary myelofibrosis. Leuk. Res. 2014, 38, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Song, M.K.; Chung, J.S.; Lim, S.N.; Lee, G.W.; Lee, S.M.; Lee, N.K.; Choi, J.C.; Oh, S.Y. Usefulness of spleen volume measured by computed tomography for predicting clinical outcome in primary myelofibrosis. Int. J. Hematol. 2016, 104, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Palandri, F.; Palumbo, G.A.; Bonifacio, M.; Tiribelli, M.; Benevolo, G.; Martino, B.; Abruzzese, E.; D’Adda, M.; Polverelli, N.; Bergamaschi, M.; et al. Baseline factors associated with response to ruxolitinib: An independent study on 408 patients with myelofibrosis. Oncotarget 2017, 8, 79073–79086. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene Mutation | Patient Characteristics | Results Associated with Splenomegaly | Reference |
|---|---|---|---|
| JAK2-V617F Homozygous mutation | 210 patients with PMF - JAK2 V617F wild type (n = 111) - JAK2 heterozygous type (n = 109) - JAK2 homozygous type (n = 84) | Patients with JAK2 V617F homozygous type → Larger splenomegaly, higher WBC count than those with wild/heterogenous type (p < 0.001) | [38] |
| CALR mutation | 617 patients with PMF. - JAK2 V617F mutation (n = 399) - CALR mutation (n = 140) - MPL mutation (n = 25) - triple negative (n = 53) | Patients with CALR mutation → longer large-splenomegaly-free survivals than remained patients (p < 0.001) | [39] |
| High risk mutations - AZXL1, EZH1, IDH1/2 | 85 MF patients treated with ruxolitinib - no mutation in AZXL1, EZH1 or IDH1/2 (n = 68) - ≥1 mutation in AZXL1, EZH1 or IDH1/2 (n = 27) | Patients with ≥ 1 mutation in AZXL1, EZH1 or IDH1/2 or those with ≥ 3 mutations of any types → significantly less likely have the response than those with no AZXL1, EZH1 or IDH1/2 (p = 0.01) or those with ≤ 2 mutation (p = 0.001) | [40] |
| Drug | Targets | Trial | Patients | Spleen Reduction Responses | References |
|---|---|---|---|---|---|
| Ruxotinib | JAK2/1/3 TYK2 | COMFORT-I (randomized phase 3) | Int-2 & high risk MF | SVR ≥ 35% at 24 weeks - 41.9% in ruxolitinib group (vs. 0.7% in BAT group) | [42] |
| COMFORT-II (randomized phase 3) | Int-2 & high risk MF | SVR ≥ 35% at 48 weeks - 28% in ruxolitinib group (vs. 0% in BAT group) | [43] | ||
| Pacritinib | JAK2/1/3 FLT3 | PERSIST-I (randomized phase 3) | Int-1, Int-2 & high risk MF | SVR ≥ 35% at 24 weeks - 19% in pacritinib group (vs. 5% in placebo) | [45] |
| PERSIST-II (randomized phase 3) | Int-1, Int-2 & high risk MF | SVR ≥ 35% at 24 weeks - 18% in pacritinib group (vs. 3% in placebo) | [46] | ||
| Momelotinib | JAK2/1/3 JNK2 CDK2 | SIMPLIFY-I (randomized phase 3) | Int-1, Int-2 & high risk MF | SVR ≥ 35% at 24 weeks - 26.9% in momelotinib group (vs. 29% ruxolitinib) | [47] |
SIMPLIFY-II (randomized phase 3) | Int-1, Int-2 & high risk MF | SVR ≥ 35% at 24 weeks - momelotinib, not superior to placebo/ruxolitinib group | [48] | ||
| Fedratinib | JAK2/1/3 TYK2 FLT3 RET | JAKARTA (randomized phase 3) | Int-2 & high risk MF | SVR ≥ 35% at 24 weeks - 36% in 400 mg fedratinib group - 40% in 500 mg fedratinib group vs. 1% in placebo group | [50] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, M.-K.; Park, B.-B.; Uhm, J.-E. Understanding Splenomegaly in Myelofibrosis: Association with Molecular Pathogenesis. Int. J. Mol. Sci. 2018, 19, 898. https://doi.org/10.3390/ijms19030898
Song M-K, Park B-B, Uhm J-E. Understanding Splenomegaly in Myelofibrosis: Association with Molecular Pathogenesis. International Journal of Molecular Sciences. 2018; 19(3):898. https://doi.org/10.3390/ijms19030898
Chicago/Turabian StyleSong, Moo-Kon, Byeong-Bae Park, and Ji-Eun Uhm. 2018. "Understanding Splenomegaly in Myelofibrosis: Association with Molecular Pathogenesis" International Journal of Molecular Sciences 19, no. 3: 898. https://doi.org/10.3390/ijms19030898
APA StyleSong, M. -K., Park, B. -B., & Uhm, J. -E. (2018). Understanding Splenomegaly in Myelofibrosis: Association with Molecular Pathogenesis. International Journal of Molecular Sciences, 19(3), 898. https://doi.org/10.3390/ijms19030898