Development and Characterisation of a Human Chronic Skin Wound Cell Line—Towards an Alternative for Animal Experimentation

 ,
,
Abstract
:1. Introduction
2. Results
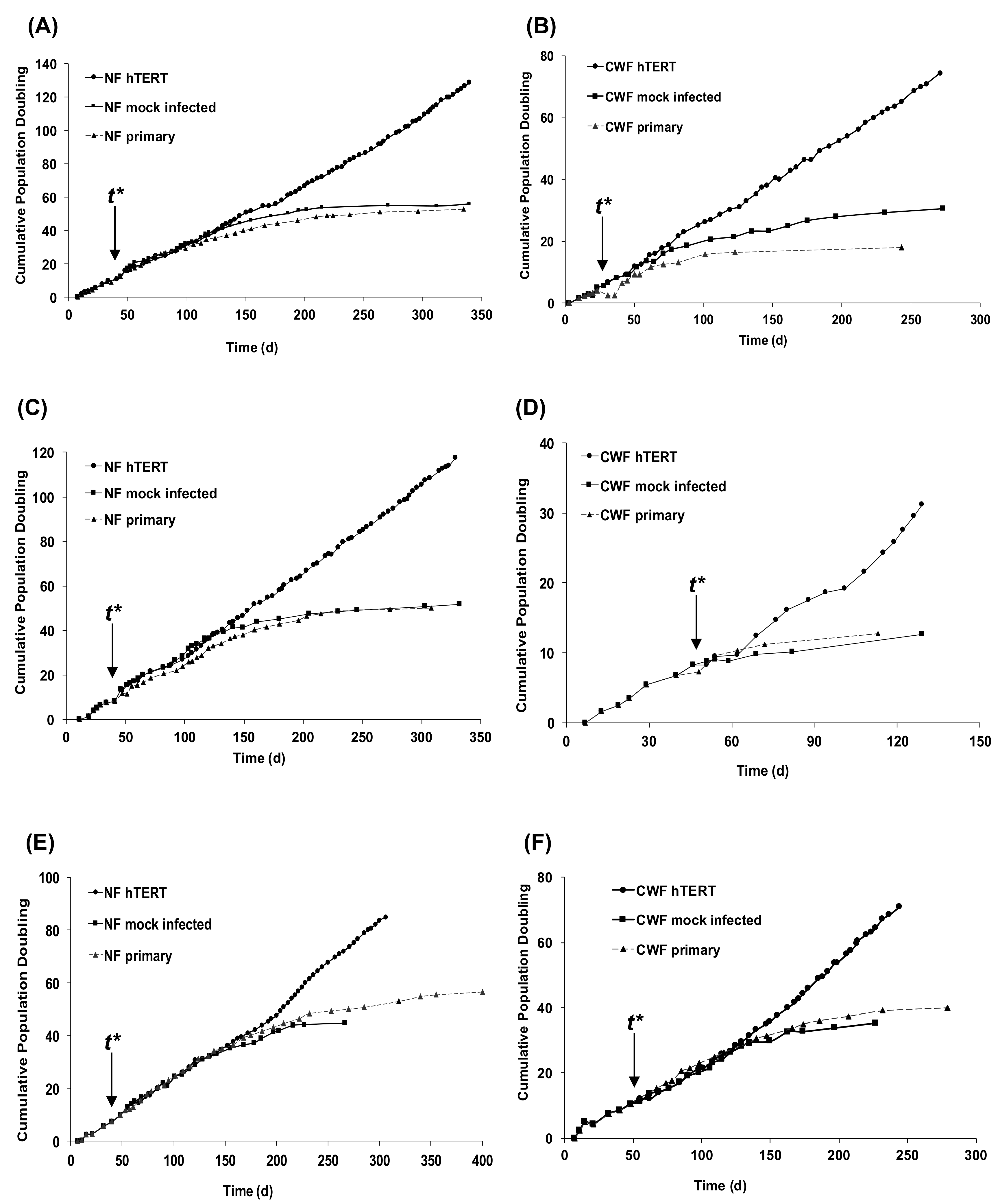
2.1. Introduction of hTERT Allows Primary NF and CWF Cell Strains to Escape Replicative Senescence
2.2. Immortalised Cell Lines Express hTERT and hTR and Have Active Telomerase
2.3. The Immortalisation Process Does Not Distort Gene Expression Signatures
2.4. Immortalisation Does Not Reverse the Disease-Specific Wound Healing Cellular Phenotype
3. Discussion
4. Materials and Methods
4.1. Patients and Tissues
4.2. Establishment of Immortalized Chronic Wound and Patient-Matched NFs
4.3. Telomerase Repeat Amplification Protocol (TRAP) Assay
4.4. Reverse Transcription Polymerase Chain Reaction
4.5. Microarray Analysis
4.6. In Vitro Wounding Studies
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Markova, A.; Mostow, E.N. US skin disease assessment: Ulcer and wound care. Dermatol. Clin. 2012, 30, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Frykberg, R.G.; Banks, J. Challenges in the treatment of chronic wounds. Adv. Wound Care 2015, 4, 560–582. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, B.M. Chronic wound care guidelines issued. JAMA 2007, 297, 938–939. [Google Scholar] [PubMed]
- Budgen, V. Evaluating the impact on patients of living with a leg ulcer. Nurs. Times 2004, 100, 30–31. [Google Scholar] [PubMed]
- Iglesias, C.P.; Birks, Y.; Nelson, E.A.; Scanlon, E.; Cullum, N.A. Quality of life of people with venous leg ulcers: A comparison of the discriminative and responsive characteristics of two generic and a disease specific instruments. Qual. Life Res. 2005, 14, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Palfreyman, S.; Michaels, J.; Brazier, J. Development of a tool to examine the effect of venous ulcers on patients’ quality of life. Nurs. Stand. 2007, 21, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Van Korlaar, I.; Vossen, C.; Rosendaal, F.; Cameron, L.; Bovill, E.; Kaptein, A. Quality of life in venous disease. Thromb. Haemost. 2003, 90, 27–35. [Google Scholar] [PubMed]
- Gunnel, R.T.; Hjelmgren, J. Annual costs of treatment for venous leg ulcers in Sweden and the united kingdom. Wound Repair Regen. 2005, 13, 13–18. [Google Scholar]
- Buemi, M.; Galeano, M.; Sturiale, A.; Ientile, R.; Crisafulli, C.; Parisi, A.; Catania, M.; Calapai, G.; Impala, P.; Aloisi, C.; et al. Recombinant human erythropoietin stimulates angiogenesis and healing of ischemic skin wounds. Shock 2004, 22, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Gould, L.J.; Leong, M.; Sonstein, J.; Wilson, S. Optimization and validation of an ischemic wound model. Wound Repair Regen. 2005, 13, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Arslantas, M.K.; Arslantas, R.; Tozan, E.N. Effects of systemic erythropoietin on ischemic wound healing in rats. Ostomy Wound Manag. 2015, 61, 28–33. [Google Scholar]
- Kloeters, O.; Jia, S.-X.; Roy, N.; Schultz, G.S.; Leinfellner, G.; Mustoe, T.A. Alteration of Smad3 signaling in ischemic rabbit dermal ulcer wounds. Wound Repair Regen. 2007, 15, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Mogford, J.E.; Liu, W.R.; Reid, R.; Chiu, C.; Said, H.; Chen, S.; Harley, C.B.; Mustoe, T.A. Adenoviral human telomerase reverse transcriptase dramatically improves ischemic wound healing without detrimental immune response in an aged rabbit model. Hum. Gene Ther. 2006, 17, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Mogford, J.E.; Sisco, M.; Bonomo, S.R.; Robinson, A.M.; Mustoe, T.A. Impact of aging on gene expression in a rat model of ischemic cutaneous wound healing1. J. Surg. Res. 2004, 118, 190–196. [Google Scholar] [CrossRef]
- Kamler, M.; Lehr, H.A.; Barker, J.H.; Saetzler, R.K.; Galla, T.J.; Messmer, K. Impact of ischemia on tissue oxygenation and wound healing: Intravital microscopic studies on the hairless mouse ear model. Eur. Surg. Res. 1993, 25, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Chien, S. Ischemic rabbit ear model created by minimally invasive surgery. Wound Repair Regen. 2007, 15, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Biswas, S.; Khanna, S.; Gordillo, G.; Bergdall, V.; Green, J.; Marsh, C.B.; Gould, L.J.; Sen, C.K. Characterization of a preclinical model of chronic ischemic wound. Physiol. Genom. 2009, 37, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, A.N.; Kesl, S.L.; Sherwood, J.; Wu, M.; Gould, L.J. Demonstration of the rat ischemic skin wound model. J. Vis. Exp. 2015, e52637. [Google Scholar] [CrossRef] [PubMed]
- Ruedrich, E.D.; Henzel, M.K.; Hausman, B.S.; Bogie, K.M. Reference gene identification for reverse transcription-quantitative polymerase chain reaction analysis in an ischemic wound-healing model. J. Biomol. Tech. 2013, 24, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Jia, S.; Tye, R.; Xu, W.; Zhong, A.; Hong, S.J.; Galiano, R.D.; Mustoe, T.A. Topical administration of oxygenated hemoglobin improved wound healing in an ischemic rabbit ear model. Plast. Reconstr. Surg. 2016, 137, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.; Wilhelmi, B.J. A simplified technique for producing an ischemic wound model. J. Vis. Exp. 2012, e3341. [Google Scholar] [CrossRef] [PubMed]
- Wall, I.B.; Moseley, R.; Baird, D.M.; Kipling, D.; Giles, P.; Laffafian, I.; Price, P.E.; Thomas, D.W.; Stephens, P. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J. Investig. Dermatol. 2008, 128, 2526–2540. [Google Scholar] [CrossRef] [PubMed]
- Agren, M.S.; Steenfos, H.H.; Dabelsteen, S.; Hansen, J.B.; Dabelsteen, E. Proliferation and mitogenic response to PDGF-BB of fibroblasts isolated from chronic venous leg ulcers is ulcer-age dependent. J. Investig. Dermatol. 1999, 112, 463–469. [Google Scholar] [PubMed]
- Peake, M.A.; Caley, M.; Giles, P.J.; Wall, I.; Enoch, S.; Davies, L.C.; Kipling, D.; Thomas, D.W.; Stephens, P. Identification of a transcriptional signature for the wound healing continuum. Wound Repair Regen. 2014, 22, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Tauzin, H.; Robin, S.; Humbert, P.; Viennet, C.; Saas, P.; Courderot-Masuyer, C.; Muret, P. Can leg ulcer fibroblasts phenotype be influenced by human amniotic membrane extract? Cell Tissue Bank. 2014, 15, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, F.; Jennewein, M.; Bubel, M.; Holstein, J.H.; Pohlemann, T.; Oberringer, M. Soft tissue fibroblasts from well healing and chronic human wounds show different rates of myofibroblasts in vitro. Mol. Biol. Rep. 2013, 40, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.-C.; Hu, M.-L. The limitations and validities of senescence associated-β-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp. Gerontol. 2005, 40, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.A.; Williamson, R.C.; Bally, B.; Cowell, C.A.; Brooks, R.; Stephens, P.; Harrison, A.J.; Bass, M.D. Ultrasonic stimulation of mouse skin reverses the healing delays in diabetes and aging by activation of Rac1. J. Investig. Dermatol. 2015, 135, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Menocal, L.; Salgado, M.; Ford, D.; Van Badiavas, E. Stimulation of skin and wound fibroblast migration by mesenchymal stem cells derived from normal donors and chronic wound patients. Stem Cells Transl. Med. 2012, 1, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, D.P.; Faria, D.T.; Nickoloff, B.J.; Poverini, P.J.; Kunkel, S.; Burdick, M.; Strieter, R.M. Chemokine and inflammatory cytokine changes during chronic wound healing. Wound Repair Regen. 1997, 5, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.J.; Yager, D.R.; Pozez, A.L.; Olutoye, O.O.; Crossland, M.C.; Diegelmann, R.F.; Cohen, I.K. Interleukin-1α and collagenase activity are elevated in chronic wounds. Plast. Reconstr. Surg. 1998, 102, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Trengove, N.J.; Bielefeldt-Ohmann, H.; Stacey, M.C. Mitogenic activity and cytokine levels in non-healing and healing chronic leg ulcers. Wound Repair Regen. 2000, 8, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Doyle, J.W.; Roth, T.P.; Dunn, R.M.; Lawrence, W.T. IL-10 and GM-CSF expression and the presence of antigen-presenting cells in chronic venous ulcers. J. Surg. Res. 1998, 79, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Ongenae, K.C.; Phillips, T.J.; Park, H.Y. Level of fibronectin mRNA is markedly increased in human chronic wounds. Dermatol. Surg. 2000, 26, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Herrick, S.E.; Sloan, P.; McGurk, M.; Freak, L.; McCollum, C.N.; Ferguson, M.W. Sequential changes in histologic pattern and extracellular matrix deposition during the healing of chronic venous ulcers. Am. J. Pathol. 1992, 141, 1085–1095. [Google Scholar] [PubMed]
- Masters, J.R. Hela cells 50 years on: The good, the bad and the ugly. Nat. Rev. Cancer 2002, 2, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Gey, G.O.; Coffman, W.D.; Kubicek, M.T. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res. 1952, 12, 264–265. [Google Scholar]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chang, E.; Cherry, A.M.; Bangs, C.D.; Oei, Y.; Bodnar, A.; Bronstein, A.; Chiu, C.-P.; Herron, G.S. Human endothelial cell life extension by telomerase expression. J. Biol. Chem. 1999, 274, 26141–26148. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Van Waarde-Verhagen, M.A.; Van Assen-Bolt, A.J.; Nieuwenhuis, B.; Rodemann, H.P.; Prowse, K.R.; Linskens, M.H. Reconstitution of active telomerase in primary human foreskin fibroblasts: Effects on proliferative characteristics and response to ionizing radiation. Int. J. Radiat. Biol. 2004, 80, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Bono, Y.; Kyo, S.; Takakura, M.; Maida, Y.; Mizumoto, Y.; Nakamura, M.; Nomura, K.; Kiyono, T.; Inoue, M. Creation of immortalised epithelial cells from ovarian endometrioma. Br. J. Cancer 2012, 106, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Kurose, M.; Kojima, T.; Koizumi, J.; Kamekura, R.; Ninomiya, T.; Murata, M.; Ichimiya, S.; Osanai, M.; Chiba, H.; Himi, T.; et al. Induction of claudins in passaged hTERT-transfected human nasal epithelial cells with an extended life span. Cell Tissue Res. 2007, 330, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.P.; Holt, S.E.; Ouellette, M.; Kaur, K.J.; Yan, Y.; Wilson, K.S.; White, M.A.; Wright, W.E.; Shay, J.W. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat. Genet. 1999, 21, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Choi, K.H.; Ouellette, M.M. Use of exogenous hTERT to immortalize primary human cells. Cytotechnology 2004, 45, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.J.; Clothier, C.; Swan, D.C.; Saretzki, G. Immediate and gradual gene expression changes in telomerase over-expressing fibroblasts. Biochem. Biophys. Res. Commun. 2010, 399, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Carney, S.A.; Tahara, H.; Swartz, C.D.; Risinger, J.I.; He, H.; Moore, A.B.; Haseman, J.K.; Barrett, J.C.; Dixon, D. Immortalization of human uterine leiomyoma and myometrial cell lines after induction of telomerase activity: Molecular and phenotypic characteristics. Lab. Investig. 2002, 82, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, C.; Hou, M.; Komurasaki, T.; Zheng, C.; Henriksson, M.; Sedivy, J.M.; Bjorkholm, M.; Teh, B.T.; Nordenskjold, M.; Xu, D. Molecular characterization of human telomerase reverse transcriptase-immortalized human fibroblasts by gene expression profiling: Activation of the epiregulin gene. Cancer Res. 2003, 63, 1743–1747. [Google Scholar] [PubMed]
- Wang, Z.; Yi, J.; Li, H.; Deng, L.F.; Tang, X.M. Extension of life-span of normal human fibroblasts by reconstitution of telomerase activity. Shi Yan Sheng Wu Xue Bao 2000, 33, 129–140. [Google Scholar] [PubMed]
- Farwell, D.G.; Shera, K.A.; Koop, J.I.; Bonnet, G.A.; Matthews, C.P.; Reuther, G.W.; Coltrera, M.D.; McDougall, J.K.; Klingelhutz, A.J. Genetic and epigenetic changes in human epithelial cells immortalized by telomerase. Am. J. Pathol. 2000, 156, 1537–1547. [Google Scholar] [CrossRef]
- Noble, J.R.; Zhong, Z.H.; Neumann, A.A.; Melki, J.R.; Clark, S.J.; Reddel, R.R. Alterations in the p16(INK4a) and p53 tumor suppressor genes of hTERT-immortalized human fibroblasts. Oncogene 2004, 23, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Pirzio, L.M.; Freulet-Marriere, M.A.; Bai, Y.; Fouladi, B.; Murnane, J.P.; Sabatier, L.; Desmaze, C. Human fibroblasts expressing hTERT show remarkable karyotype stability even after exposure to ionizing radiation. Cytogenet. Genome Res. 2004, 104, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Milyavsky, M.; Shats, I.; Erez, N.; Tang, X.; Senderovich, S.; Meerson, A.; Tabach, Y.; Goldfinger, N.; Ginsberg, D.; Harris, C.C.; et al. Prolonged culture of telomerase-immortalized human fibroblasts leads to a premalignant phenotype. Cancer Res. 2003, 63, 7147–7157. [Google Scholar] [PubMed]
- Cristofalo, V.J.; Allen, R.G.; Pignolo, R.J.; Martin, B.G.; Beck, J.C. Relationship between donor age and the replicative lifespan of human cells in culture: A reevaluation. Proc. Natl. Acad. Sci. USA 1998, 95, 10614–10619. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Wu, F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res. 1997, 25, 2595–2597. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multilpe testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Days | NF Primary Patient 1 | CWF Primary Patient 1 | NF Mock Patient 1 | CWF Mock Patient 1 | NF hTERT Patient 1 | CWF hTERT Patient 1 |
| 0–50 | 2.660395186 | 1.372377017 | 2.903460321 | 1.676095021 | 2.820859002 | 1.761228228 |
| 50–100 | 2.063483514 | 1.155055922 | 2.21362413 | 1.491181981 | 2.17190845 | 2.350465441 |
| 100–150 | 1.708425038 | 0.238615271 | 1.931265023 | 0.808847164 | 2.888996473 | 2.100595582 |
| 150–200 | 1.092771348 | - | 1.209523855 | 0.589792188 | 2.546683912 | 2.147975214 |
| 200–250 | 0.65051411 | - | 0.306199448 | - | 2.911358566 | 2.340073042 |
| 250–300 | - | - | - | - | 3.386106828 | 1.914658417 |
| 300–350 | - | - | - | - | 4.037277255 | 2.316985136 |
| Days | NF Primary Patient 2 | CWF Primary Patient 2 | NF Mock Patient 2 | CWF Mock Patient 2 | NF hTERT Patient 2 | CWF hTERT Patient 2 |
| 0–50 | 2.00003978 | 1.430818888 | 2.702882371 | 1.408288338 | 2.702421121 | 1.315226122 |
| 50–100 | 1.897508302 | 0.642582236 | 2.506688459 | 0.389073493 | 2.084423303 | 1.526944077 |
| 100–150 | 2.048256328 | - | 1.937150809 | - | 3.091048229 | 2.73604129 |
| 150–200 | 1.362063393 | - | 0.741986217 | - | 2.848323718 | 2.544191407 |
| 200–250 | 0.468038965 | - | - | - | 2.975591664 | 3.613521307 |
| 250–300 | 0.225106176 | - | - | - | 3.282693337 | 3.238706858 |
| 300–350 | - | - | - | - | 3.283963285 | 3.136433714 |
| Days | NF Primary Patient 3 | CWF Primary Patient 3 | NF Mock Patient 3 | CWF Mock Patient 3 | NF hTERT Patient 3 | CWF hTERT Patient 3 |
| 0–50 | 1.773899657 | 1.838731796 | 1.876174641 | 1.811924471 | 1.864799274 | 1.957757805 |
| 50–100 | 2.137331544 | 1.820469606 | 2.227068604 | 1.765460977 | 2.175212065 | 1.931044873 |
| 100–150 | 1.700143374 | 1.313762689 | 1.821538813 | 1.463326161 | 2.040014915 | 1.948431103 |
| 150–200 | 1.359040254 | 1.018979963 | 1.30906374 | 0.656850067 | 1.866400168 | 1.991061535 |
| 200–250 | 0.96345786 | 0.381403418 | 0.386622971 | 0.60575738 | 3.009158197 | 1.847844344 |
| 250–300 | 0.494600255 | - | - | - | 2.453037313 | 1.455326622 |
| 300–350 | 0.918257283 | - | - | - | 2.36463098 | 1.521344782 |
| Normal (NF) | Wound (CWF) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 h | 6 h after Serum Stimulation | 0 h | 6 h after Serum Stimulation | |||||
| Early | FDR 0.05 | 0 | FDR 0.05 | 0 | FDR 0.05 | 0 | FDR 0.05 | 0 |
| Mid | FDR 0.05 | 0 | FDR 0.05 | 6 | FDR 0.05 | 0 | FDR 0.05 | 0 |
| Late | FDR 0.05 | 3 | FDR 0.05 | 0 | FDR 0.05 | 0 | FDR 0.05 | 0 |
| Normal (NF) | Wound (CWF) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 h | 6 h after Serum Stimulation | 0 h | 6 h after Serum Stimulation | |||||
| Early | FDR 0.01 | 476 | FDR 0.01 | 193 | FDR 0.01 | 161 | FDR 0.01 | 145 |
| Mid | FDR 0.01 | 653 | FDR 0.01 | 866 | FDR 0.01 | 292 | FDR 0.01 | 203 |
| Late | FDR 0.01 | 418 | FDR 0.01 | 507 | FDR 0.01 | 446 | FDR 0.01 | 660 |
| Pathway | Genes |
|---|---|
| DNA replication | TOP2A, MCM3, RRM2, MCM6, NASP, CDC6, RFC3, CDK2, FEN1, CHEK1, POLE2, GINS1, TERT, MCM7, NF2, CIZ1, GMNN, DTL, PDGFC, MCM4 |
| DNA strand elongation involved in DNA replication | MCM3, MCM6, RFC3, FEN1, GINS1, MCM7, MCM4 |
| cell cycle | TOP2A, MCM3, RANBP2, RRM2, MCM6, NASP, TUBB3, GAS7, CCNA2, TFDP2, FGFR2, RARA, MAPRE3, CDC6, ZWINT, RFC3, CKS2, CDK2, RASSF1, FEN1, BARD1, CHEK1, POLE2, GINS1, TUBB2C, HMG20B, SMAD6, TGFB2, TPX2, MCM7, KIF2C, TUBB, CDKN2C, ARAP1, CDKN1C, CKAP2, GMNN, CEP55, DTL, MCM4, RACGAP1 |
| cell cycle phase | POLD2, TOP2A, PSMD3, MCM3, RRM2, CHMP1A, TUBB3, DNM2, RANBP1, UBE2S, RFC5, CDK1, CAV2, MAD2L1, CCNA2, BUB1B, DLGAP5, CDC6, ZWINT, TRIP13, AURKA, CKS2, DBF4, CDK2, PSMB9, GTSE1, CDC7, KIF23, FEN1, CCNF, CENPA, GINS1, AKT1, DGKZ, TUBB2C, TUBB, ID4, BUB1, CDKN3, HGF, TPX2, CDKN2D, SSNA1, MCM7, KIF2C, KPNA2, RANGAP1, TFDP1, NEK1, TCF3, MDM2, PRC1, NUSAP1, GMNN, CEP55, PBK, CDKN1C, CENPN, NCAPG2, TBRG4, CCDC99, MCM4 |
| organelle fission | CHMP1A, TUBB3, RANBP1, CDK1, CAV2, MAD2L1, CCNA2, BUB1B, DLGAP5, CDC6, ZWINT, AURKA, CDK2, KIF23, CCNF, CENPA, TUBB, BUB1, HGF, TPX2, KIF2C, RANGAP1, NEK1, NUSAP1, CEP55, PBK, CENPN, NCAPG2, CCDC99 |
| mitotic cell cycle | POLD2, TOP2A, PSMD3, MCM3, RRM2, CHMP1A, TUBB3, DNM2, RANBP1, UBE2S, RFC5, CDK1, CAV2, MAD2L1, CCNA2, BUB1B, DLGAP5, CDC6, ZWINT, AURKA, DBF4, CDK2, PSMB9, GTSE1, CDC7, KIF23, FEN1, CCNF, CENPA, GINS1, AKT1, DGKZ, TUBB2C, TUBB, ID4, BUB1, CDKN3, HGF, TPX2, CDKN2D, SSNA1, MCM7, KIF2C, KPNA2, RANGAP1, TFDP1, NEK1, TCF3, MDM2, PRC1, NUSAP1, GMNN, CEP55, PBK, CDKN1C, CENPN, NCAPG2, TBRG4, CCDC99, MCM4 |
| cell division | PPP1CA, CHMP1A, UBE2S, CDK1, MAD2L1, CCNA2, FGFR2, BUB1B, CDC6, ZWINT, AURKA, CKS2, CDK2, CDC7, KIF23, CCNF, FGF1, FGF5, MDK, PTN, BUB1, TPX2, KIF2C, NEK1, PRC1, NUSAP1, CEP55, PDGFC, PDGFD, NCAPG2, CCDC99, RACGAP1 |
| mitosis | CHMP1A, TUBB3, RANBP1, CDK1, CAV2, MAD2L1, CCNA2, BUB1B, DLGAP5, CDC6, ZWINT, AURKA, CDK2, KIF23, CCNF, CENPA, TUBB, BUB1, HGF, TPX2, KIF2C, RANGAP1, NEK1, NUSAP1, CEP55, PBK, CENPN, NCAPG2, CCDC99 |
| M phase | TOP2A, CHMP1A, TUBB3, RANBP1, UBE2S, CDK1, CAV2, MAD2L1, CCNA2, BUB1B, DLGAP5, CDC6, ZWINT, TRIP13, AURKA, CKS2, CDK2, KIF23, CCNF, CENPA, TUBB, BUB1, HGF, TPX2, KIF2C, KPNA2, RANGAP1, NEK1, PRC1, NUSAP1, CEP55, PBK, CENPN, NCAPG2, CCDC99 |
| DNA replication | RFC2, POLD2, PCNA, TOP2A, RRM1, RPA1, MCM3, MCM5, RRM2, MCM6, NASP, MCM2, TK1, TYMS, RNASEH2A, RFC5, CDC6, RFC4, RFC3, DBF4, POLA2, FEN1, CHAF1B, POLA1, RAD51, CCNE2, CHEK1, PDGFA, CIZ1, GINS1, TERT, CENPF, MCM7, DUT, CDT1, NF2, ORC5, MCM4, CDC34, GMNN, DTL, RMI1, TIPIN, MCM10, GINS2 |
| cell cycle checkpoint | RFC2, TOP2A, BUB3, RPA1, MCM3, MCM5, MCM6, BIRC5, MCM2, CCNB2, CDC20, UBE2C, RFC5, CDK1, MAD2L1, CCNA2, FANCG, BUB1B, CDC6, RFC4, ZWINT, RFC3, DBF4, PSMB9, GTSE1, TTK, POLA1, CCNE2, CHEK1, CENPF, MCM7, BUB1, CDT1, ORC5, MCM4, CCNB1, ZWILCH, DTL, TIPIN, MCM10 |
| cell division | PPP1CA, BUB3, NCAPD2, CDC25B, PSRC1, BIRC5, CCNB2, CDC20, UBE2C, CDK1, MAD2L1, CCNA2, FGFR2, BUB1B, CDC6, ZWINT, AURKA, NDC80, CKS2, SMC2, KIF11, NEK2, KIF23, CCNF, CCNE2, RAB35, PDGFA, DIAPH2, CENPF, MAEA, FGF5, MDK, KIF2C, AURKB, PTN, BUB1, TGFB2, TPX2, OIP5, CCNB1, PRC1, NUSAP1, ZWILCH, CEP55, FBXO5, TIPIN, NCAPG2, ERCC6L, ASPM, CDCA3, CDCA8, RACGAP1 |
| S phase of mitotic cell cycle | RFC2, POLD2, PCNA, RPA1, MCM3, MCM5, MCM6, MCM2, BCL6, RFC5, CDC6, RFC4, RFC3, PSMB9, POLA2, FEN1, POLA1, GINS1, MCM7, CDT1, ORC5, MCM4, GINS2 |
| telomere maintenance via recombination | RFC2, POLD2, PCNA, RPA1, RFC5, RFC4, RFC3, POLA2, FEN1, POLA1 |
| telomere maintenance via semi-conservative replication | RFC2, POLD2, PCNA, RPA1, RFC5, RFC4, RFC3, POLA2, FEN1, POLA1 |
| M/G1 transition of mitotic cell cycle | RPA1, MCM3, MCM5, MCM6, MCM2, CDC6, DBF4, PSMB9, POLA2, POLA1, MCM7, CDT1, ORC5, MCM4, GMNN, MCM10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caley, M.; Wall, I.B.; Peake, M.; Kipling, D.; Giles, P.; Thomas, D.W.; Stephens, P. Development and Characterisation of a Human Chronic Skin Wound Cell Line—Towards an Alternative for Animal Experimentation. Int. J. Mol. Sci. 2018, 19, 1001. https://doi.org/10.3390/ijms19041001
Caley M, Wall IB, Peake M, Kipling D, Giles P, Thomas DW, Stephens P. Development and Characterisation of a Human Chronic Skin Wound Cell Line—Towards an Alternative for Animal Experimentation. International Journal of Molecular Sciences. 2018; 19(4):1001. https://doi.org/10.3390/ijms19041001
Chicago/Turabian StyleCaley, Matthew, Ivan B. Wall, Matthew Peake, David Kipling, Peter Giles, David W. Thomas, and Phil Stephens. 2018. "Development and Characterisation of a Human Chronic Skin Wound Cell Line—Towards an Alternative for Animal Experimentation" International Journal of Molecular Sciences 19, no. 4: 1001. https://doi.org/10.3390/ijms19041001
APA StyleCaley, M., Wall, I. B., Peake, M., Kipling, D., Giles, P., Thomas, D. W., & Stephens, P. (2018). Development and Characterisation of a Human Chronic Skin Wound Cell Line—Towards an Alternative for Animal Experimentation. International Journal of Molecular Sciences, 19(4), 1001. https://doi.org/10.3390/ijms19041001