Different Natural Selection Pressures on the atpF Gene in Evergreen Sclerophyllous and Deciduous Oak Species: Evidence from Comparative Analysis of the Complete Chloroplast Genome of Quercus aquifolioides with Other Oak Species


Abstract
:1. Introduction
2. Results and Discussion
2.1. Chloroplast Genome Organization in Q. aquifolioides
2.2. Repeat Sequence Analysis and Simple Sequence Repeats (SSR)
2.3. Comparison of the cp Genomes of Q. aquifolioides and Three Related Quercus Species
2.4. Genome Sequence Divergence among Quercus Species
2.5. Selection Events in Protein Coding Genes
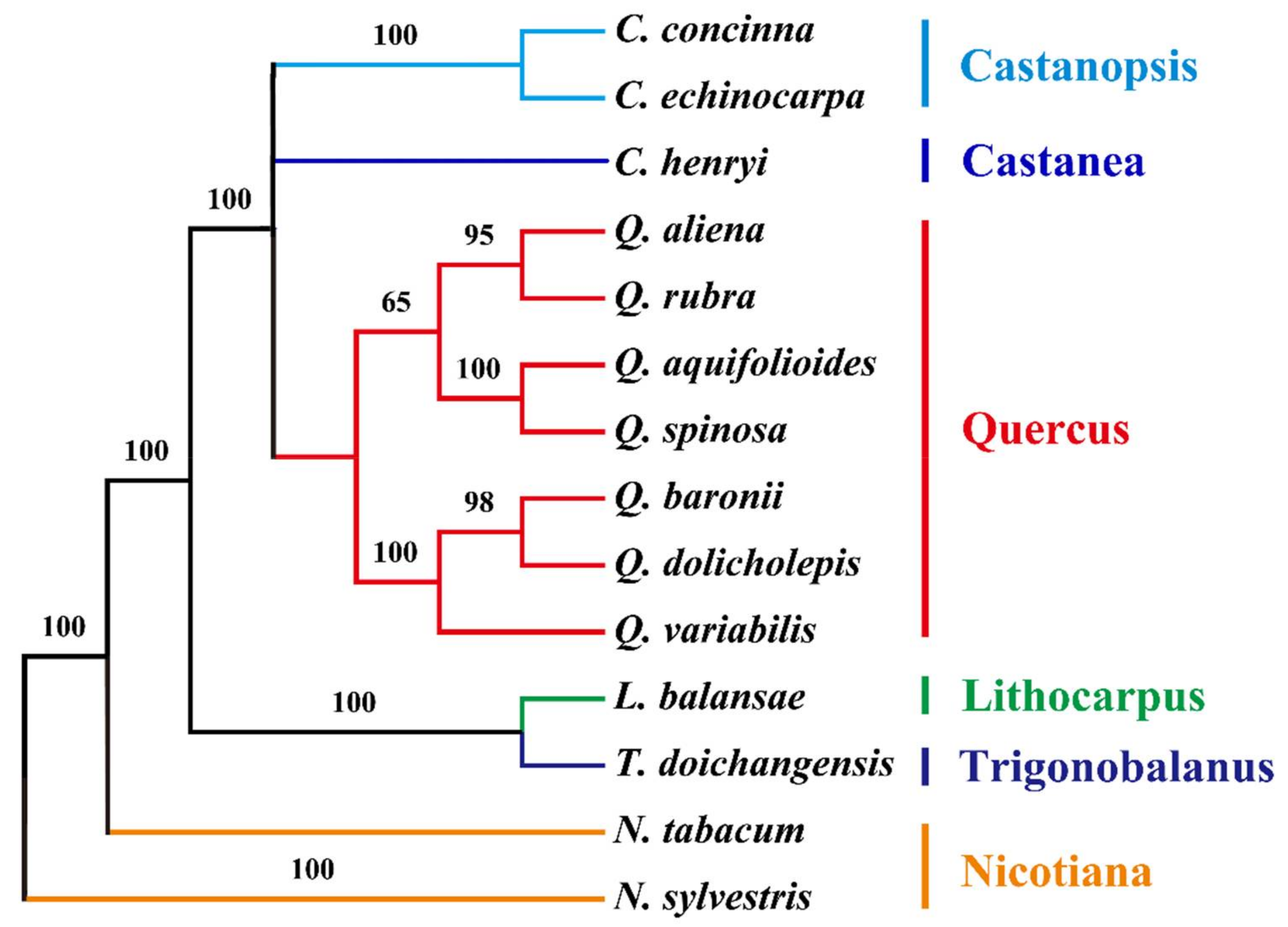
2.6. Phylogenetic Analysis of the cp Genomes of Q. aquifolioides and Related Quercus Species
3. Materials and Methods
3.1. Plant Material
3.2. Chloroplast Isolation, DNA Extraction, and Sequencing
3.3. Chloroplast Genome Assembly
3.4. Genome Annotation
3.5. Repeat Analysis
3.6. CCT Map
3.7. Sequence Divergence Analysis
3.8. Selection Pressure Analysis
3.9. Phylogenetic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McCauley, D.E.; Stevens, J.E.; Peroni, P.A.; Raveill, J.A. The spatial distribution of chloroplast DNA and allozyme polymorphisms within a population of Silene alba (Caryophyllaceae). Am. J. Bot. 1996, 83, 727–731. [Google Scholar] [CrossRef]
- Yurina, N.P.; Odintsova, M.S. Comparative structural organization of plant chloroplast and mitochondrial genomes. Genetika 1998, 34, 5–22. [Google Scholar]
- Nixon, K.C. The genus Quercus in Mexico. In Biological Diversity of Mexico: Origins and Distribution; Oxford University Press: New York, NY, USA, 1993; pp. 447–458. ISBN 019506674X. [Google Scholar]
- Keator, G.; Bazel, S. The Life of an Oak: An Intimate Portrait; Heyday Books: Berkeley, CA, USA; California Oak Foundation: Oakland, CA, USA, 1998; p. 256. ISBN 9780930588984. [Google Scholar]
- Nixon, K.C. Global and neotropical distribution and diversity of oak (genus Quercus) and oak forests. In Ecology and Conservation of Neotropical Montane Oak Forests; Springer: Berlin/Heidelberg, Germany, 2006; pp. 3–13. ISBN 364206695X. [Google Scholar]
- Trelease, W. The American oaks. Mem. Natl. Acad. Sci. 1924, 20, 1–255. [Google Scholar]
- Palmer, E.J. Hybrid oaks of North America. J. Arnold Arbor. 1948, 29, 1–48. [Google Scholar]
- Muller, C.H. Ecological control of hybridization in Quercus: A factor in the mechanism of evolution. Evolution 1952, 6, 147–161. [Google Scholar]
- Tucker, J.M. Studies in the Quercus undulata complex. I. A preliminary statement. Am. J. Bot. 1961, 48, 202–208. [Google Scholar] [CrossRef]
- Hardin, J.W. Hybridization and introgression in Quercus alba. J. Arnold Arbor. 1975, 56, 336–363. [Google Scholar]
- Rushton, B.S. Natural hybridization within the genus Quercus. Ann. For. Sci. 1993, 50, 73s–90s. [Google Scholar] [CrossRef]
- Spellenberg, R. On the hybrid nature of Quercus basaseachicensis (Fagaceae, sect. Quercus). SIDA Contrib. Bot. 1995, 16, 427–437. [Google Scholar]
- Bacilieri, R.; Ducousso, A.; Petit, R.J.; Kremer, A. Mating system and asymmetric hybridization in a mixed stand of European oaks. Evolution 1996, 50, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.J.; Preszler, R.W.; Williams, J.; Fenchel, S.; Boecklen, W.J. How discrete are oak species? Insights from a hybrid zone between Quercus grisea and Quercus gambelii. Evolution 1997, 51, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Manos, P.S.; Doyle, J.J.; Nixon, K.C. Phylogeny, biogeography, and processes of molecular differentiation in Quercus subgenus Quercus (Fagaceae). Mol. Phylogenet. Evol. 1999, 12, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Manos, P.S.; Stanford, A.M. The historical biogeography of Fagaceae: Tracking the tertiary history of temperate and subtropical forests of the Northern Hemisphere. Int. J. Plant Sci. 2001, 162, S77–S93. [Google Scholar] [CrossRef]
- Grímsson, F.; Zetter, R.; Grimm, G.W.; Pedersen, G.K.; Pedersen, A.K.; Denk, T. Fagaceae pollen from the early Cenozoic of West Greenland: Revisiting Engler’s and Chaney’s Arcto-Tertiary hypotheses. Plant Syst. Evol. 2015, 301, 809–832. [Google Scholar] [CrossRef] [PubMed]
- Simeone, M.C.; Grimm, G.W.; Papini, A.; Vessella, F.; Cardoni, S.; Tordoni, E.; Piredda, R.; Franc, A.; Denk, T. Plastome data reveal multiple geographic origins of Quercus Group Ilex. PeerJ 2016, 4, e1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denk, T.; Grimm, G.W.; Manos, P.S.; Deng, M.; Hipp, A.L. An updated infrageneric classification of the oaks: Review of previous taxonomic schemes and synthesis of evolutionary patterns. In Oaks Physiological Ecology. Exploring the Functional Diversity of Genus Quercus L.; Gil-Pelegrín, E., Peguero-Pina, J., Sancho-Knapik, D., Eds.; Springer: Cham, Switzerland, 2017; pp. 13–38. ISBN 978-3-319-69098-8. [Google Scholar]
- Huang, C.J.; Zhang, Y.T.; Bartholomew, B. Fagaeeae. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China, 1999; pp. 370–380. ISBN 0915279703. [Google Scholar]
- Du, F.K.; Hou, M.; Wang, W.; Mao, K.S.; Hampe, A. Phylogeography of Quercus aquifolioides provides novel insights into the Neogene history of a major global hotspot of plant diversity in southwest China. J. Biogeogr. 2017, 44, 294–307. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; dePamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Method Enzymol. 2005, 395, 348–384. [Google Scholar]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; Pabón-Peña, C.R.; Freimer, N.B. Compound microsatellite repeats: Practical and theoretical features. Genome Res. 1999, 9, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Kaittanis, C.; Saski, C.; Lee, S.B.; Tomkins, J.; Alverson, A.J.; Daniell, H. Phylogenetic analyses of Vitis (Vitaceae) based on complete chloroplast genome sequences: Effects of taxon sampling and phylogenetic methods on resolving relationships among rosids. BMC Evol. Biol. 2006, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Ruhlman, T.; Lee, S.B.; Jansen, R.K.; Hostetler, J.B.; Tallon, L.J.; Town, C.D.; Daniell, H. Complete plastid genome sequence of Daucus carota: Implications for biotechnology and phylogeny of angiosperms. BMC Genom. 2006, 7, 222. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.R.; Diaz, Y.C.; Penha, H.A.; Pinheiro, D.G.; Fernandes, C.C.; Miranda, V.F.; Todd, P.; Michael, T.P.; Varani, A.M. The Chloroplast Genome of Utricularia reniformis Sheds Light on the Evolution of the ndh Gene Complex of Terrestrial Carnivorous Plants from the Lentibulariaceae Family. PLoS ONE 2016, 11, e0165176. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Li, R.; Worth, J.R.; Li, X.; Li, P.; Cameron, K.M.; Fu, C.X. The Complete Chloroplast Genome of Chinese Bayberry (Morella rubra, Myricaceae): Implications for Understanding the Evolution of Fagales. Front. Plant Sci. 2017, 8, 968. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Goulding, S.E.; Wolfe, K.H.; Olmstead, R.G.; Morden, C.W. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996, 252, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Cheng, T.; Zhou, S. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 2013, 8, e77965. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Li, P.; Qiu, Y. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2016, 7, 2054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Liu, A.; Chen, J.; Wu, L.; Hu, W.; Zhang, W.; Kim, K.; Lee, S.C.; Tae-Jin Yang, T.J.; et al. The Complete Chloroplast Genome Sequences of Five Epimedium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huo, N.; Dong, L.; Wang, Y.; Zhang, S.; Young, H.A.; Feng, X.; Gu, Y.Q. Complete Chloroplast Genome Sequences of Mongolia Medicine Artemisia frigida and Phylogenetic Relationships with Other Plants. PLoS ONE 2013, 8, e57533. [Google Scholar] [CrossRef] [PubMed]
- Nazareno, A.G.; Carlsen, M.; Lohmann, L.G. Complete Chloroplast Genome of Tanaecium tetragonolobum: The First Bignoniaceae Plastome. PLoS ONE 2015, 10, e0129930. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.; Fahmy, R.M.; Mosa, K.A.; Helmy, M.; El-Feky, F.A. Identification of effective DNA barcodes for Triticum plants through chloroplast genome-wide analysis. Comput. Biol. Chem. 2017, 71, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. The neutral theory of molecular evolution and the world view of the neutralists. Genome 1983, 31, 24–31. [Google Scholar] [CrossRef]
- Hudson, G.S.; Mason, J.G. The chloroplast genes encoding subunits of the H+-ATP synthase. In Molecular Biology of Photosynthesis; Govindjee, Ed.; Springer: Dordrecht, The Netherlands, 1988; pp. 565–582. ISBN 978-94-010-7517-6. [Google Scholar]
- Cummings, M.P.; King, L.M.; Kellogg, E.A. Slipped-strand mispairing in a plastid gene: RpoC2 in grasses (Poaceae). Mol. Biol. Evol. 1994, 11, 1–8. [Google Scholar] [PubMed]
- Aldrich, P.R.; Cavender-Bares, J. Quercus. In Wild Crop Relatives: Genomic and Breeding Resources; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 89–129. ISBN 978-3-642-21249-9. [Google Scholar]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; dePamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Li, D.Z.; Li, H.T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Res. 2014, 14, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Du, F.K.; Lang, T.; Lu, S.; Wang, Y.; Li, J.; Yin, K. An improved method for chloroplast genome sequencing in non-model forest tree species. Tree Genet. Genomes 2015, 11, 114. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [PubMed]
- Stern, A.; Doron-Faigenboim, A.; Erez, E.; Martz, E.; Bacharach, E.; Pupko, T. Selecton 2007: Advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 2007, 35, W506–W511. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (and Other Methods). Version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | Sect. Heterobalanus | Sect. Lobatae | Sect. Quercus | |
|---|---|---|---|---|
| Q. aquifolioides | Q. spinosa | Q. rubra | Q. aliena | |
| Genome size/GC content | 160,415/37.0 | 160,825/36.9 | 161,304/36.8 | 160,921/36.9 |
| Coding genes: number/size | 80(7)/80,270 | 80(7)/80,812 | 80(7)/80,946 | 80(7)/80,052 |
| tRNA: number/size | 39/10,625 | 38/11,402 | 39/10,756 | 39/10,753 |
| rRNA: number/size | 8/9048 | 8/9050 | 8/9050 | 8/9048 |
| LSC: size/percent/GC content | 89,807/56/34.8 | 90,371/56.2/34.7 | 90,541/56.1/34.7 | 90,258/56.1/34.7 |
| SSC: size/percent/GC content | 18,894/11.8/31.2 | 18,732/11.6/31.2 | 19,023/56.1/30.9 | 18,972/11.8/31.0 |
| IR: size/percent/GC content | 51,754/32.2/42.7 | 51,722/32.2/47.2 | 52,740/32.7/42.7 | 51,682/32.1/42.7 |
| Introns: size/percent | 20,473/12.8 | 19,757/12.3 | 20,217/12.5 | 20,014/12.4 |
| Intergenic spacer: size/percent | 49,548/31.0 | 50,207/31.2 | 47,473/29.4 | 47,304/29.3 |
| Gene | NULL (M8a) | POSITIVE (M8) | Putative Sites under Positive Selection |
|---|---|---|---|
| rpl2 | −1177.05 | −1174.54 | 1 (131 S) |
| ycf2 | −9154.8 | −9154.87 | 5 (96 K, 932 W, 1174 P, 1291 W, 2007 R) |
| rps7 | −615.83 | −615.616 | 1 (130 E) |
| ndhD | −2145.31 | −2141.84 | 8 (170 T, 188 G, 200 L, 206 A, 362 R, 375 P, 413 Q, 504 F) |
| ycf1 | −8057.78 | −8051.12 | 8 (426 F, 529 L, 757 L, 761 L, 1007 I, 1490 Q, 1491 G, 1492 F) |
| rpoC2 | −5814.68 | −5814.8 | 26 (33 H, 131 P, 280 L, 364 I, 505 H, 542 E, 587 E, 595 P, 598 V, 626 N, 643 K, 691 G, 697 T, 815 Y, 849 G, 856 H, 898 D, 947 S, 1013 K, 1074 I, 1081 A, 1132 E, 1176 I, 1273 C, 1374 D, 1394 N) |
| rpoC1 | −2857.37 | −2855.15 | 1 (145 Y) |
| psaB | −2999.97 | −2998.62 | 3 (145 L, 238 E, 239 K) |
| ndhJ | −633.793 | −633.569 | 1 (107 A) |
| ndhC | −828.789 | −828.761 | 2 (68 V, 86 F) |
| rpl36 | −145.376 | −145.135 | 1 (20 R) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, K.; Zhang, Y.; Li, Y.; Du, F.K. Different Natural Selection Pressures on the atpF Gene in Evergreen Sclerophyllous and Deciduous Oak Species: Evidence from Comparative Analysis of the Complete Chloroplast Genome of Quercus aquifolioides with Other Oak Species. Int. J. Mol. Sci. 2018, 19, 1042. https://doi.org/10.3390/ijms19041042
Yin K, Zhang Y, Li Y, Du FK. Different Natural Selection Pressures on the atpF Gene in Evergreen Sclerophyllous and Deciduous Oak Species: Evidence from Comparative Analysis of the Complete Chloroplast Genome of Quercus aquifolioides with Other Oak Species. International Journal of Molecular Sciences. 2018; 19(4):1042. https://doi.org/10.3390/ijms19041042
Chicago/Turabian StyleYin, Kangquan, Yue Zhang, Yuejuan Li, and Fang K. Du. 2018. "Different Natural Selection Pressures on the atpF Gene in Evergreen Sclerophyllous and Deciduous Oak Species: Evidence from Comparative Analysis of the Complete Chloroplast Genome of Quercus aquifolioides with Other Oak Species" International Journal of Molecular Sciences 19, no. 4: 1042. https://doi.org/10.3390/ijms19041042
APA StyleYin, K., Zhang, Y., Li, Y., & Du, F. K. (2018). Different Natural Selection Pressures on the atpF Gene in Evergreen Sclerophyllous and Deciduous Oak Species: Evidence from Comparative Analysis of the Complete Chloroplast Genome of Quercus aquifolioides with Other Oak Species. International Journal of Molecular Sciences, 19(4), 1042. https://doi.org/10.3390/ijms19041042