PPAR Agonists and Metabolic Syndrome: An Established Role?



 and
and 
Abstract
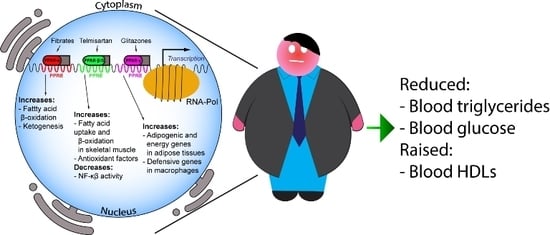
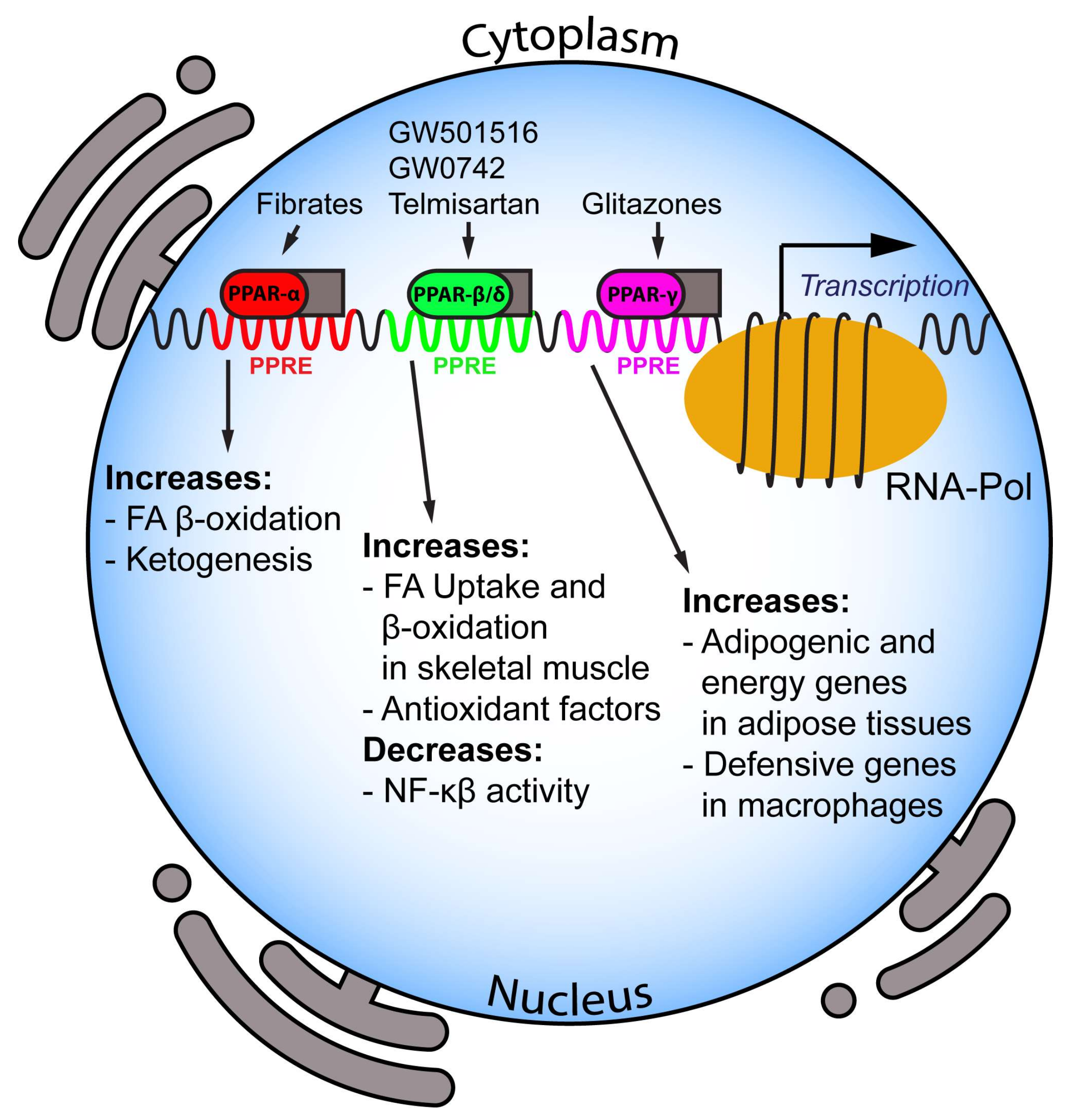
:
1. Introduction
2. Peroxisome Proliferator-Activated Receptor: Key Players in Energy Homeostasis
2.1. PPAR-α
2.2. PPAR-β/δ
2.3. PPAR-γ
3. PPAR-α: Fibrates and Omega-3 Fatty Acids in the Metabolic Syndrome
3.1. Fibrates
Fibrates: Evidence from the Most Recent Clinical Trials
3.2. Omega-3
Omega-3: Evidence from the Most Recent Clinical Trials
4. PPAR-γ Agonists
5. PPAR Dual Agonists
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Carvalho Vidigal, F.; Bressan, J.; Babio, N.; Salas-Salvado, J. Prevalence of metabolic syndrome in Brazilian adults: A systematic review. BMC Public Health 2013, 13, 1198. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, W.; Lun, Z.; Zhang, H.; Sun, Z.; Kanu, J.S.; Qiu, S.; Cheng, Y.; Liu, Y. Prevalence of metabolic syndrome in Mainland China: A meta-analysis of published studies. BMC Public Health 2016, 16, 296. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the metabolic syndrome in the United States, 2003-2012. JAMA 2015, 313, 1973–1974. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [PubMed]
- National Cholesterol Education Program (NCEP); Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar]
- Einhorn, D.; Reaven, G.M.; Cobin, R.H.; Ford, E.; Ganda, O.P.; Handelsman, Y.; Hellman, R.; Jellinger, P.S.; Kendall, D.; Krauss, R.M.; et al. American College of Endocrinology position statement on the insulin resistance syndrome. Endocr. Pract. 2003, 9, 237–252. [Google Scholar] [PubMed]
- Alberti, K.G.; Zimmet, P.; Shaw, J.; IDF Epidemiology Task Force Consensus Group. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Ferri, N.; Ruscica, M. Proprotein convertase subtilisin/kexin type 9 (PCSK9) and metabolic syndrome: Insights on insulin resistance, inflammation, and atherogenic dyslipidemia. Endocrine 2016, 54, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Fisman, E.Z.; Motro, M.; Adler, Y. Atherogenic dyslipidemia in metabolic syndrome and type 2 diabetes: Therapeutic options beyond statins. Cardiovasc. Diabetol. 2006, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J.; Redfern, J.S.; McGovern, M.E.; Giral, P. Niacin and fibrates in atherogenic dyslipidemia: Pharmacotherapy to reduce cardiovascular risk. Pharmacol. Ther. 2010, 126, 314–345. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M. The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. Prog. Mol. Biol. Transl. Sci. 2014, 121, 217–266. [Google Scholar] [PubMed]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Bortolini, M.; Wright, M.B.; Bopst, M.; Balas, B. Examining the safety of PPAR agonists—Current trends and future prospects. Expert Opin. Drug Saf. 2013, 12, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Corsini, A.; Bellosta, S.; Davidson, M.H. Pharmacokinetic interactions between statins and fibrates. Am. J. Cardiol. 2005, 96, 44K–49K; discussion 34K–35K. [Google Scholar] [CrossRef] [PubMed]
- Magni, P.; Macchi, C.; Morlotti, B.; Sirtori, C.R.; Ruscica, M. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur. J. Intern. Med. 2015, 26, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, G. PPARs Integrate the Mammalian Clock and Energy Metabolism. PPAR Res. 2014, 2014, 653017. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Staels, B.; Auwerx, J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim. Biophys. Acta 1996, 1302, 93–109. [Google Scholar] [CrossRef]
- Moore, K.J.; Rosen, E.D.; Fitzgerald, M.L.; Randow, F.; Andersson, L.P.; Altshuler, D.; Milstone, D.S.; Mortensen, R.M.; Spiegelman, B.M.; Freeman, M.W. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat. Med. 2001, 7, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Saluja, I.; Granneman, J.G.; Skoff, R.P. PPAR delta agonists stimulate oligodendrocyte differentiation in tissue culture. Glia 2001, 33, 191–204. [Google Scholar] [CrossRef]
- Cermenati, G.; Audano, M.; Giatti, S.; Carozzi, V.; Porretta-Serapiglia, C.; Pettinato, E.; Ferri, C.; D’Antonio, M.; De Fabiani, E.; Crestani, M.; et al. Lack of sterol regulatory element binding factor-1c imposes glial Fatty Acid utilization leading to peripheral neuropathy. Cell Metab. 2015, 21, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar] [PubMed]
- Riserus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Kim, M.; Park, H.S.; Kim, H.S.; Jeon, M.J.; Oh, K.S.; Koh, E.H.; Won, J.C.; Kim, M.S.; Oh, G.T.; et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem. Biophys. Res. Commun. 2006, 340, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Grefhorst, A.; van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.J. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J. Biol. Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Peters, J.M.; Harris, R.A. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem. Biophys. Res. Commun. 2001, 287, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Keller, H.; Wahli, W. Peroxisome proliferator-activated receptors A link between endocrinology and nutrition? Trends Endocrinol. Metab. 1993, 4, 291–296. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, R.; Gervois, P.P.; Verschuren, L.; Staels, B.; Princen, H.M.; Kooistra, T. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFkappa B-C/EBP-beta complex formation. Blood 2003, 101, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.R.; Clarke, S.; Rodgers, K.; Thornhill, B.; Peters, J.M.; Gonzalez, F.J.; Gimble, J.M. Effect of peroxisome proliferator-activated receptor alpha activators on tumor necrosis factor expression in mice during endotoxemia. Infect. Immun. 1999, 67, 3488–3493. [Google Scholar] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Amri, E.Z.; Bonino, F.; Ailhaud, G.; Abumrad, N.A.; Grimaldi, P.A. Cloning of a protein that mediates transcriptional effects of fatty acids in preadipocytes. Homology to peroxisome proliferator-activated receptors. J. Biol. Chem. 1995, 270, 2367–2371. [Google Scholar] [CrossRef] [PubMed]
- Hertz, R.; Berman, I.; Keppler, D.; Bar-Tana, J. Activation of gene transcription by prostacyclin analogues is mediated by the peroxisome-proliferators-activated receptor (PPAR). Eur. J. Biochem. 1996, 235, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Holst, D.; Luquet, S.; Nogueira, V.; Kristiansen, K.; Leverve, X.; Grimaldi, P.A. Nutritional regulation and role of peroxisome proliferator-activated receptor delta in fatty acid catabolism in skeletal muscle. Biochim. Biophys. Acta 2003, 1633, 43–50. [Google Scholar] [CrossRef]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, K.S.; Billin, A.N. PPARbeta/delta ligands as modulators of the inflammatory response. Curr. Opin. Investig. Drugs 2008, 9, 463–469. [Google Scholar] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, Y.; Tang, Z.; Zhang, H.; Qin, X.; Zhu, Y.; Guan, Y.; Wang, X.; Staels, B.; Chien, S.; et al. Suppression of pro-inflammatory adhesion molecules by PPAR-delta in human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Zingarelli, B.; Piraino, G.; Hake, P.W.; O’Connor, M.; Denenberg, A.; Fan, H.; Cook, J.A. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am. J. Pathol. 2010, 177, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARgamma in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Zhang, X.; Zhu, C.; Tang, X.; Yu, F.; Shang, G.W.; Cai, X. Molecular Mechanisms of PPAR-gamma Governing MSC Osteogenic and Adipogenic Differentiation. Curr. Stem Cell Res. Ther. 2016, 11, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Yoshioka, S.; Yoshioka, T.; Ushiyama, I.; Horikoshi, H. Characterization of new oral antidiabetic agent CS-045. Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes 1988, 37, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J. Clin. Investig. 2000, 106, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Miles, P.D.; Barak, Y.; He, W.; Evans, R.M.; Olefsky, J.M. Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J. Clin. Investig. 2000, 105, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamaki, J.; Mykkanen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S.; et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Steger, D.J.; Zhuo, D.; Qatanani, M.; Mullican, S.E.; Tuteja, G.; Manduchi, E.; Grant, G.R.; Lazar, M.A. Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol. Cell. Biol. 2010, 30, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Foryst-Ludwig, A.; Hartge, M.; Clemenz, M.; Sprang, C.; Hess, K.; Marx, N.; Unger, T.; Kintscher, U. PPARgamma activation attenuates T-lymphocyte-dependent inflammation of adipose tissue and development of insulin resistance in obese mice. Cardiovasc. Diabetol. 2010, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Motoki, T.; Kurobe, H.; Hirata, Y.; Nakayama, T.; Kinoshita, H.; Rocco, K.A.; Sogabe, H.; Hori, T.; Sata, M.; Kitagawa, T. PPAR-gamma agonist attenuates inflammation in aortic aneurysm patients. Gen. Thorac. Cardiovasc. Surg. 2015, 63, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Ginty, M.; Kemp, K.; Scolding, N.; Wilkins, A. The PPAR-gamma agonist pioglitazone protects cortical neurons from inflammatory mediators via improvement in peroxisomal function. J. Neuroinflammation 2012, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.; Castellino, G.; Banach, M.; Toth, P.P.; Ivanova, E.; Orekhov, A.N.; Montalto, G.; Rizzo, M. PPAR Agonists, Atherogenic Dyslipidemia and Cardiovascular Risk. Curr. Pharm. Des. 2017, 23, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Dash, S.; Morgantini, C.; Hegele, R.A.; Lewis, G.F. Pharmacological Targeting of the Atherogenic Dyslipidemia Complex: The Next Frontier in CVD Prevention Beyond Lowering LDL Cholesterol. Diabetes 2016, 65, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.A.; Kizhakepunnur, L.G.; Bahekar, A.; Arora, R.R. The role of fibrates in the prevention of cardiovascular disease—A pooled meta-analysis of long-term randomized placebo-controlled clinical trials. Am. Heart J. 2007, 154, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Sirtori, C.R. Mechanisms of action of absorbable hypolipidemic drugs. Adv. Exp. Med. Biol. 1985, 183, 241–252. [Google Scholar] [PubMed]
- Sirtori, C.R. The FIELD study. Lancet 2006, 367, 1141–1142; author reply 1142–1143. [Google Scholar] [CrossRef]
- Elam, M.B.; Ginsberg, H.N.; Lovato, L.C.; Corson, M.; Largay, J.; Leiter, L.A.; Lopez, C.; O’Connor, P.J.; Sweeney, M.E.; Weiss, D.; et al. Association of Fenofibrate Therapy With Long-term Cardiovascular Risk in Statin-Treated Patients With Type 2 Diabetes. JAMA Cardiol. 2017, 2, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Corsini, A.; Sirtori, C.; Ruscica, M. PPAR-alpha agonists are still on the rise: An update on clinical and experimental findings. Expert Opin. Investig. Drugs 2017, 26, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Simental-Mendia, L.E.; Simental-Mendia, M.; Sanchez-Garcia, A.; Banach, M.; Atkin, S.L.; Gotto, A.M., Jr.; Sahebkar, A. Effect of fibrates on glycemic parameters: A systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol. Res. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Vamecq, J.; Latruffe, N. Medical significance of peroxisome proliferator-activated receptors. Lancet 1999, 354, 141–148. [Google Scholar] [CrossRef]
- Brown, J.D.; Plutzky, J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation 2007, 115, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Flory, J.H.; Ellenberg, S.; Szapary, P.O.; Strom, B.L.; Hennessy, S. Antidiabetic action of bezafibrate in a large observational database. Diabetes Care 2009, 32, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Raza-Iqbal, S.; Tanaka, T.; Anai, M.; Inagaki, T.; Matsumura, Y.; Ikeda, K.; Taguchi, A.; Gonzalez, F.J.; Sakai, J.; Kodama, T. Transcriptome Analysis of K-877 (a Novel Selective PPARalpha Modulator (SPPARMalpha))-Regulated Genes in Primary Human Hepatocytes and the Mouse Liver. J. Atheroscler. Thromb. 2015, 22, 754–772. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T.; Nakamura, J.; Maegawa, H.; Yoshioka, N.; Tanizawa, Y.; et al. Effects of Pemafibrate, a Novel Selective PPARalpha Modulator, on Lipid and Glucose Metabolism in Patients With Type 2 Diabetes and Hypertriglyceridemia: A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial. Diabetes Care 2018, 41, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendia, L.E.; Mikhailidis, D.P.; Pirro, M.; Banach, M.; Sirtori, C.R.; Ruscica, M.; Reiner, Z. Effect of statin therapy on plasma apolipoprotein CIII concentrations: A systematic review and meta-analysis of randomized controlled trials. J. Clin. Lipidol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S.; Group, K.S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor alpha modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and Safety of Pemafibrate Versus Fenofibrate in Patients with High Triglyceride and Low HDL Cholesterol Levels: A Multicenter, Placebo-Controlled, Double-Blind, Randomized Trial. J. Atheroscler. Thromb. 2018. [Google Scholar] [CrossRef] [PubMed]
- Camejo, G. Phase 2 clinical trials with K-877 (pemafibrate): A promising selective PPAR-alpha modulator for treatment of combined dyslipidemia. Atherosclerosis 2017, 261, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and safety of K-877, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), in combination with statin treatment: Two randomised, double-blind, placebo-controlled clinical trials in patients with dyslipidaemia. Atherosclerosis 2017, 261, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Handb. Exp. Pharmacol. 2015, 224, 3–51. [Google Scholar] [PubMed]
- Fruchart, J.C. Pemafibrate (K-877), a novel selective peroxisome proliferator-activated receptor alpha modulator for management of atherogenic dyslipidaemia. Cardiovasc. Diabetol. 2017, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- NCT03071692. Pemafibrate to Reduce Cardiovascular OutcoMes by Reducing Triglycerides IN patiENts with diabeTes (PROMINENT). Available online: https://www.clinicaltrials.gov/ct2/results?cond=&term=NCT03071692&cntry=&state=&city=&dist= (accessed on 13 April 2018).
- Sirtori, C.R.; Galli, C.; Franceschini, G. Fraudulent (and non fraudulent) fatty acids for human health. Eur. J. Clin. Investig. 1993, 23, 686–689. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Mechanisms of action of (n-3) fatty acids. J. Nutr. 2012, 142, 592S–599S. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jia, Y.; Fu, T.; Viswakarma, N.; Bai, L.; Rao, M.S.; Zhu, Y.; Borensztajn, J.; Reddy, J.K. Sustained activation of PPARalpha by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J. 2012, 26, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L. Update on the management of severe hypertriglyceridemia—Focus on free fatty acid forms of omega-3. Drug Des. Dev. Ther. 2015, 9, 2129–2137. [Google Scholar]
- Pirillo, A.; Catapano, A.L. Omega-3 polyunsaturated fatty acids in the treatment of hypertriglyceridaemia. Int. J. Cardiol. 2013, 170 (Suppl. S1), S16–S20. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.F.; Burke, F.M.; Soffer, D.E. Review of Cardiometabolic Effects of Prescription Omega-3 Fatty Acids. Curr. Atheroscler. Rep. 2017, 19, 60. [Google Scholar] [CrossRef] [PubMed]
- Sperling, L.S.; Nelson, J.R. History and future of omega-3 fatty acids in cardiovascular disease. Curr. Med. Res. Opin. 2016, 32, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Pizzini, A.; Lunger, L.; Demetz, E.; Hilbe, R.; Weiss, G.; Ebenbichler, C.; Tancevski, I. The Role of Omega-3 Fatty Acids in Reverse Cholesterol Transport: A Review. Nutrients 2017, 9, 1099. [Google Scholar] [CrossRef] [PubMed]
- Pahlavani, M.; Ramalho, T.; Koboziev, I.; LeMieux, M.J.; Jayarathne, S.; Ramalingam, L.; Filgueiras, L.R.; Moustaid-Moussa, N. Adipose tissue inflammation in insulin resistance: Review of mechanisms mediating anti-inflammatory effects of omega-3 polyunsaturated fatty acids. J. Investig. Med. 2017, 65, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Kuda, O.; Rossmeisl, M.; Kopecky, J. Omega-3 fatty acids and adipose tissue biology. Mol. Aspects Med. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.U.; Ohmori, K.; Hashimoto, T.; Kamitori, K.; Yamaguchi, F.; Noma, T.; Igarashi, J.; Tsuboi, K.; Tokuda, M.; Nishiyama, A.; et al. GPR120 in adipocytes has differential roles in the production of pro-inflammatory adipocytokines. Biochem. Biophys. Res. Commun. 2017, 486, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Bargut, T.C.; Silva-e-Silva, A.C.; Souza-Mello, V.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Mice fed fish oil diet and upregulation of brown adipose tissue thermogenic markers. Eur. J. Nutr. 2016, 55, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Lopez, T.; Cereijo, R.; Turatsinze, J.V.; Planavila, A.; Cairo, M.; Gavalda-Navarro, A.; Peyrou, M.; Moure, R.; Iglesias, R.; Giralt, M.; et al. The lipid sensor GPR120 promotes brown fat activation and FGF21 release from adipocytes. Nat. Commun. 2016, 7, 13479. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, A.J.; Cruz, A.L.; Heyward, W.L.; Bulkow, L.R.; Hall, D.; Barstaed, L.; Connor, W.E. Elevated concentrations of plasma omega-3 polyunsaturated fatty acids among Alaskan Eskimos. Am. J. Clin. Nutr. 1994, 59, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Park, H.G.; Zhang, J.Y.; Lawrence, P.; Liu, G.; Subramanian, N.; Kothapalli, K.S.; Brenna, J.T. Brown but not white adipose cells synthesize omega-3 docosahexaenoic acid in culture. Prostaglandins Leukot. Essent. Fatty Acids 2016, 104, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Baragetti, A.; Catapano, A.L.; Norata, G.D. Translating the biology of adipokines in atherosclerosis and cardiovascular diseases: Gaps and open questions. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Monk, J.M.; Liddle, D.M.; De Boer, A.A.; Brown, M.J.; Power, K.A.; Ma, D.W.; Robinson, L.E. Fish-oil-derived n-3 PUFAs reduce inflammatory and chemotactic adipokine-mediated cross-talk between co-cultured murine splenic CD8+ T cells and adipocytes. J. Nutr. 2015, 145, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Ballantyne, C.M.; Braeckman, R.A.; Stirtan, W.G.; Doyle, R.T., Jr.; Philip, S.; Soni, P.N.; Juliano, R.A. Icosapent Ethyl (Eicosapentaenoic Acid Ethyl Ester): Effects Upon High-Sensitivity C-Reactive Protein and Lipid Parameters in Patients With Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2015, 13, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Buoite Stella, A.; Gortan Cappellari, G.; Barazzoni, R.; Zanetti, M. Update on the Impact of Omega 3 Fatty Acids on Inflammation, Insulin Resistance and Sarcopenia: A Review. Int. J. Mol. Sci. 2018, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.Y.; Jacobson, T.A. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: A systematic review and meta-analysis. Curr. Atheroscler. Rep. 2011, 13, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, J. Omega-3 Fatty Acid Formulations in Cardiovascular Disease: Dietary Supplements are Not Substitutes for Prescription Products. Am. J. Cardiovasc. Drugs 2016, 16, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.S.G.; Susekov, A.V.; de Bruin, T.W.A.; Kvarnstrom, M.; Yang, H.; Davidson, M.H. Omega-3 carboxylic acids in patients with severe hypertriglyceridemia: EVOLVE II, a randomized, placebo-controlled trial. J. Clin. Lipidol. 2018, 12, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.; Maki, K.C.; Susekov, A.; Ezhov, M.; Nordestgaard, B.G.; Machielse, B.N.; Kling, D.; Davidson, M.H. Omega-3 free fatty acids for the treatment of severe hypertriglyceridemia: The EpanoVa fOr Lowering Very high triglyceridEs (EVOLVE) trial. J. Clin. Lipidol. 2014, 8, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.C.; Orloff, D.G.; Nicholls, S.J.; Dunbar, R.L.; Roth, E.M.; Curcio, D.; Johnson, J.; Kling, D.; Davidson, M.H. A highly bioavailable omega-3 free fatty acid formulation improves the cardiovascular risk profile in high-risk, statin-treated patients with residual hypertriglyceridemia (the ESPRIT trial). Clin. Ther. 2013, 35, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet 1999, 354, 447–455. [Google Scholar]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Kromhout, D.; Giltay, E.J.; Geleijnse, J.M.; Alpha Omega Trial Group. n-3 fatty acids and cardiovascular events after myocardial infarction. N. Engl. J. Med. 2010, 363, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.; Halsey, J.; Kromhout, D.; Gerstein, H.C.; Marchioli, R.; Tavazzi, L.; Geleijnse, J.M.; Rauch, B.; Ness, A.; Galan, P.; et al. Associations of Omega-3 Fatty Acid Supplement Use With Cardiovascular Disease Risks: Meta-analysis of 10 Trials Involving 77917 Individuals. JAMA Cardiol. 2018, 3, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Steg, P.G.; Brinton, E.A.; Jacobson, T.A.; Miller, M.; Tardif, J.C.; Ketchum, S.B.; Doyle, R.T., Jr.; Murphy, S.A.; Soni, P.N.; et al. Rationale and design of REDUCE-IT: Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial. Clin. Cardiol. 2017, 40, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Baldessin, L.; Boccia, D.; Racagni, G.; Mitro, N. Non-insulin anti-diabetic drugs: An update on pharmacological interactions. Pharmacol. Res. 2017, 115, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K. Rosiglitazone revisited: An updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch. Intern. Med. 2010, 170, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Wolski, K.; Nicholls, S.J.; Nissen, S.E. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: A meta-analysis of randomized trials. JAMA 2007, 298, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. CHICAGO, PERISCOPE and PROactive: CV risk modification in diabetes with pioglitazone. Fundam. Clin. Pharmacol. 2009, 23, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Nicholls, S.J.; Wolski, K.; Nesto, R.; Kupfer, S.; Perez, A.; Jure, H.; De Larochelliere, R.; Staniloae, C.S.; Mavromatis, K.; et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: The PERISCOPE randomized controlled trial. JAMA 2008, 299, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Tuzcu, E.M.; Wolski, K.; Bayturan, O.; Lavoie, A.; Uno, K.; Kupfer, S.; Perez, A.; Nesto, R.; Nissen, S.E. Lowering the triglyceride/high-density lipoprotein cholesterol ratio is associated with the beneficial impact of pioglitazone on progression of coronary atherosclerosis in diabetic patients: Insights from the PERISCOPE (Pioglitazone Effect on Regression of Intravascular Sonographic Coronary Obstruction Prospective Evaluation) study. J. Am. Coll. Cardiol. 2011, 57, 153–159. [Google Scholar] [PubMed]
- Mazzone, T.; Meyer, P.M.; Feinstein, S.B.; Davidson, M.H.; Kondos, G.T.; D’Agostino, R.B., Sr.; Perez, A.; Provost, J.C.; Haffner, S.M. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: A randomized trial. JAMA 2006, 296, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.; Meyer, P.M.; Haffner, S.; Feinstein, S.; D’Agostino, R., Sr.; Kondos, G.T.; Perez, A.; Chen, Z.; Mazzone, T. Increased high-density lipoprotein cholesterol predicts the pioglitazone-mediated reduction of carotid intima-media thickness progression in patients with type 2 diabetes mellitus. Circulation 2008, 117, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Kernan, W.N.; Viscoli, C.M.; Furie, K.L.; Young, L.H.; Inzucchi, S.E.; Gorman, M.; Guarino, P.D.; Lovejoy, A.M.; Peduzzi, P.N.; Conwit, R.; et al. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2016, 374, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Viscoli, C.M.; Young, L.H.; Furie, K.L.; Gorman, M.; Lovejoy, A.M.; Dagogo-Jack, S.; Ismail-Beigi, F.; Korytkowski, M.T.; Pratley, R.E.; et al. Pioglitazone Prevents Diabetes in Patients With Insulin Resistance and Cerebrovascular Disease. Diabetes Care 2016, 39, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Viscoli, C.M.; Inzucchi, S.E.; Young, L.H.; Insogna, K.L.; Conwit, R.; Furie, K.L.; Gorman, M.; Kelly, M.A.; Lovejoy, A.M.; Kernan, W.N.; et al. Pioglitazone and Risk for Bone Fracture: Safety Data from a Randomized Clinical Trial. J. Clin. Endocrinol. Metab. 2017, 102, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dougherty, E.J.; Danner, R.L. PPARgamma signaling and emerging opportunities for improved therapeutics. Pharmacol. Res. 2016, 111, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Pena, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vazquez-Carrera, M. PPARbeta/delta: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Chew, G.T.; Watts, G.F. New peroxisome proliferator-activated receptor agonists: Potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin. Pharmacother. 2014, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Majumder, A.; Ray, S. Observational study of effects of Saroglitazar on glycaemic and lipid parameters on Indian patients with type 2 diabetes. Sci. Rep. 2015, 5, 7706. [Google Scholar] [CrossRef] [PubMed]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Jani, R.H.; Pai, V.; Jha, P.; Jariwala, G.; Mukhopadhyay, S.; Bhansali, A.; Joshi, S. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI). Diabetes Technol. Ther. 2014, 16, 63–71. [Google Scholar] [PubMed]
- Balakumar, P.; Rose, M.; Ganti, S.S.; Krishan, P.; Singh, M. PPAR dual agonists: Are they opening Pandora’s Box? Pharmacol. Res. 2007, 56, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Pavanello, C.; Calabresi, L.; Ruscica, M. Nutraceutical approaches to metabolic syndrome. Ann. Med. 2017, 49, 678–697. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Unmet need for primary prevention in individuals with hypertriglyceridaemia not eligible for statin therapy according to European Society of Cardiology/European Atherosclerosis Society guidelines: A contemporary population-based study. Eur. Heart J. 2018, 39, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Corsini, A.; Sirtori, C.R.; Ruscica, M. Present therapeutic role of cholesteryl ester transfer protein inhibitors. Pharmacol. Res. 2018, 128, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.M.; Nordestgaard, B.G. Is It Time for New Thinking about High-Density Lipoprotein? Arterioscler. Thromb. Vasc. Biol. 2018, 38, 484–486. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Value | Alternative Indicator | |
|---|---|---|
| Waist circumference | * >94 cm in males, >80 cm in females ** >102 cm in males, >88 cm in females | |
| Raised blood pressure | Systolic ≥130 and/or diastolic ≥85 mm Hg | Treatment of previously diagnosed hypertension |
| Raised FPG | ≥100 mg/dL (5.6 mmol/L) | Previously diagnosed of T2DM |
| Raised TG | >150 mg/dL (1.7 mmol/L) | Specific pharmacological treatment |
| Reduced HDL-C | <40 mg/dL (1.0 mmol/L) in males <50 mg/dL (1.3 mmol/L) in females | Specific pharmacological treatment |
| PPAR-α Agonist | Clinic Study | Major Findings |
|---|---|---|
| Pemafibrate | Phase 3 (JapicCTI-142412; clinicaltrials.jp) follow-up: 24 weeks subjects: 166 [74] | 1. Reduction in TGs: −45% 2. Decrement in non-HDL 3. Increase in HDL cholesterol |
| Phase 3 (JapicCTI-142620; clinicaltrials.jp) follow-up: 24 weeks subjects: 225 [76] | 1. Reduction in TGs: −46.2% 2. A further −6.5% TG reduction compared to fenofibrate | |
| Phase 3 (JapicCTI-121764; clinicaltrials.jp) follow-up: 12 weeks subjects: 489 [77] | 1. TGs: −46.3% (0.1 mg/day), −46.7% (0.2 mg/day) and −51.8% (0.4 mg/day) vs. −38.3% (fenofibrate 100 mg/day) and −51.5% (fenofibrate 200 mg/day) | |
| Phase 2 follow-up: 12 weeks subjects: 188 [79] | 1. Reduction in TGs: range from −46.1% to −53.4% | |
| Phase 2 follow-up: 24 weeks subjects: 423 [79] | 1. Reduction in TGs: range from −46.8% to −50.8% | |
| On going Phase 3 trial PROMINENT (Pemafibrate to Reduce Cardiovascular OutcoMes by Reducing Triglycerides IN patiENts with diabeTes)—NCT03071692 | Outcomes: First occurrence of nonfatal myocardial infarction, nonfatal ischemic stroke, hospitalization for unstable angina requiring unplanned coronary revascularization, and CV death. | |
| Omega-3 | EVOLVEII (Epanova® for Lowering Very High Triglycerides II)—NCT02009865 Phase 3 follow-up: 12 weeks subjects: 162 [106] | 1. Reduction in TGs: −14.2% 2. Reduction in non-HDL-C: −9% |
| EVOLVE (The EpanoVa fOr Lowering Very high triglyceridEs)—NCT01242527 Phase 3 follow-up: 12 weeks subjects: 399 [107] | 1. Reduction in TGs: range −25.5%/−30.9% 2. Reduction in non-HDL-C: range from −6.9% to −9.6% | |
| ESPRIT (EPANOVA Combined with a STATIN in PATIENTS With HYPERTRIGLYCERIDEMIA to Reduce Non-HDL CHOLESTEROL)—NCT01408303. Phase 3 follow-up: 6 weeks subjects: 647 [108] | 1. Reduction in TGs: range from −14.6% to −20.6% 2. Reduction in non-HDL-C: range from −3.9% to −6.9% | |
| On going phase 3 trials: (i) STRENGTH (Study to assess statin residual risk Reduction with Epanova in high cardiovascular risk patients with Hypertriglyceridemia)—NCT02104817 (ii) REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention)—NCT01492361 [113] | Outcomes: First occurrence of cardiovascular death, nonfatal MI, nonfatal stroke, emergent/elective coronary revascularization, or hospitalization for unstable angina | |
| Pioglitazone | The PERISCOPE Trial (Pioglitazone Effect on Regression of Intravascular Sonographic Coronary Obstruction Prospective Evaluation)—NCT00225277 Phase 3 follow-up: 18 months subjects: 543 [119] | 1. Percent atheroma volume change: −0.16% 2. Raise in HDL-C: +5.7 mg/dL 3. Decrement in TGs: −16.3 mg/dL |
| The CHICAGO (Carotid Intima-Media Thickness in Atherosclerosis Using Pioglitazone) trial—NCT00225264 Phase 3 follow-up: 72 weeks subjects: 462 [121,122] | 1. Progression of mean CIMT: −0.013 mm vs. glimepiride 2. Progression of maximum CIMT: −0.024 mm vs. glimepiride 3. HDL-C: +14% | |
| The IRIS (Insulin Resistance Intervention after Stroke)—NCT00091949 Phase 3 follow-up: 4.8 years subjects: 3876 [123,124] | 1. Reduction of stroke or MI in insulin resistant patients 2. Reduction in recurrence of diabetes: −52% | |
| Elafibranor | GOLDEN trial—NCT01694849 Phase 2b follow-up: 52 weeks subjects: 256 [131] | 1. NASH resolution in 19% of patients |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. https://doi.org/10.3390/ijms19041197
Botta M, Audano M, Sahebkar A, Sirtori CR, Mitro N, Ruscica M. PPAR Agonists and Metabolic Syndrome: An Established Role? International Journal of Molecular Sciences. 2018; 19(4):1197. https://doi.org/10.3390/ijms19041197
Chicago/Turabian StyleBotta, Margherita, Matteo Audano, Amirhossein Sahebkar, Cesare R. Sirtori, Nico Mitro, and Massimiliano Ruscica. 2018. "PPAR Agonists and Metabolic Syndrome: An Established Role?" International Journal of Molecular Sciences 19, no. 4: 1197. https://doi.org/10.3390/ijms19041197
APA StyleBotta, M., Audano, M., Sahebkar, A., Sirtori, C. R., Mitro, N., & Ruscica, M. (2018). PPAR Agonists and Metabolic Syndrome: An Established Role? International Journal of Molecular Sciences, 19(4), 1197. https://doi.org/10.3390/ijms19041197