Identification of Dysregulated microRNA Networks in Schwann Cell-Like Cultures Exposed to Immune Challenge: Potential Crosstalk with the Protective VIP/PACAP Neuropeptide System

 , ,
, , 
Abstract
:
1. Introduction
2. Results
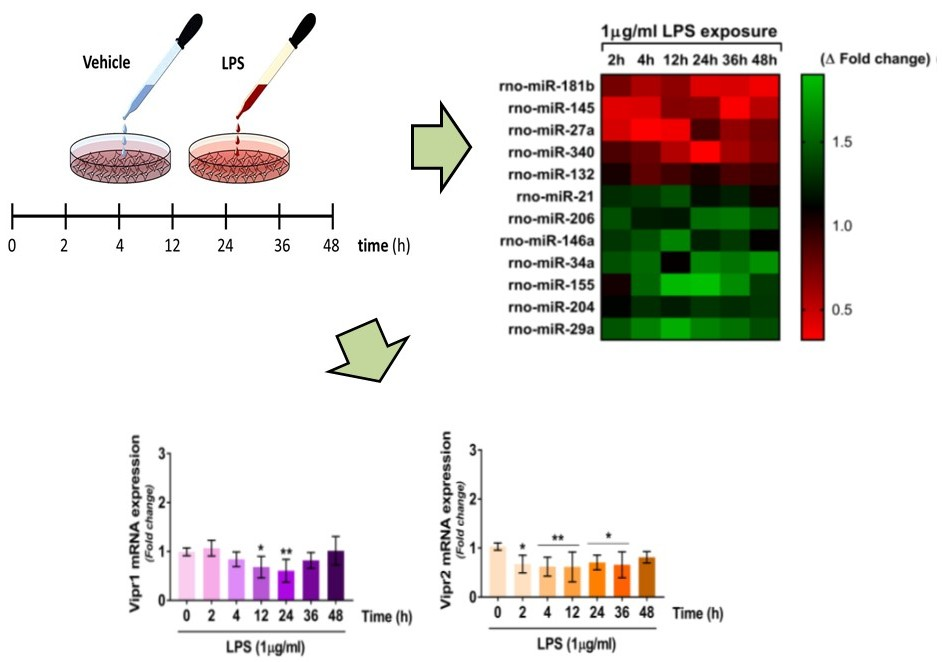
2.1. Effects of LPS Treatment on RT4 SCs Viability
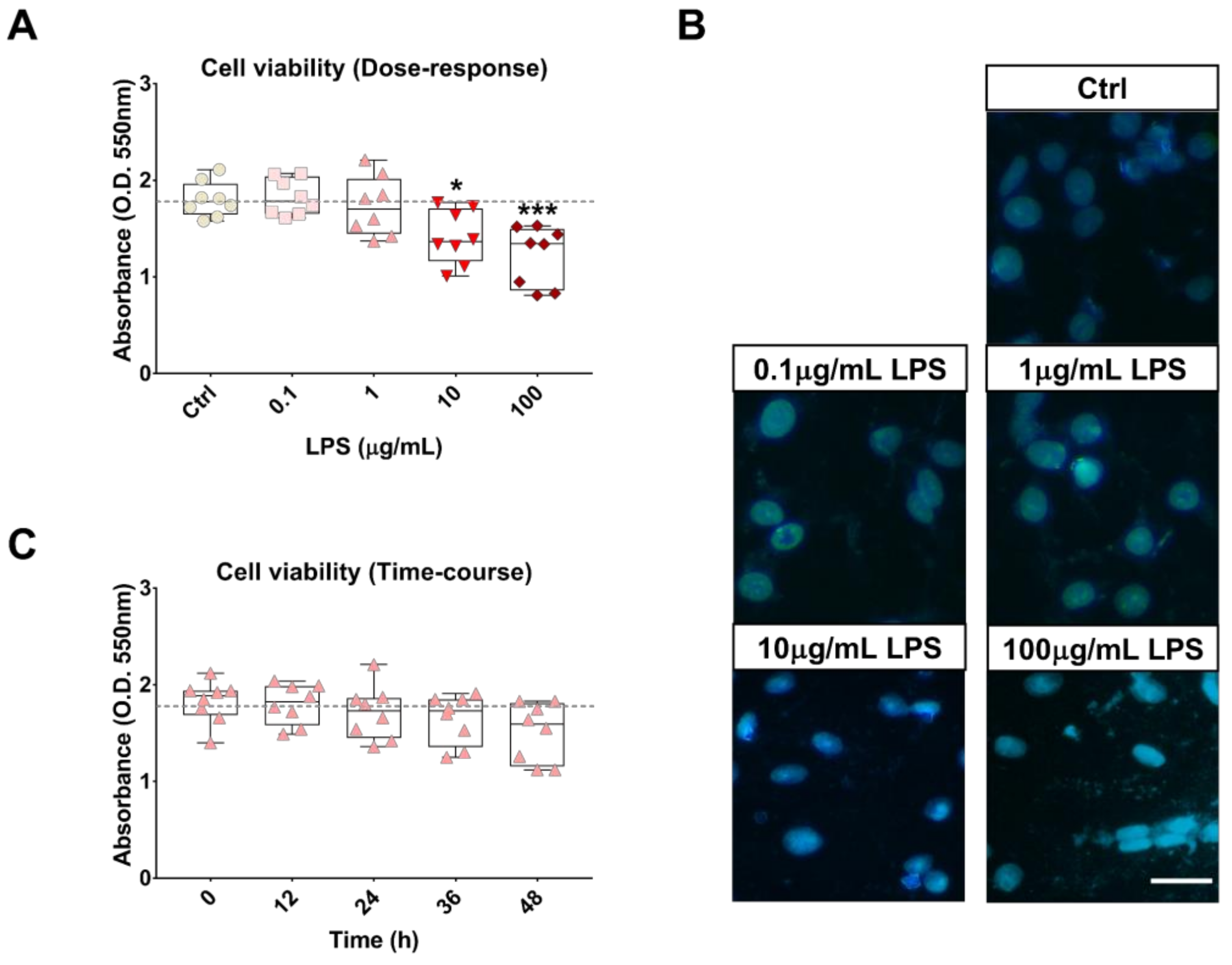
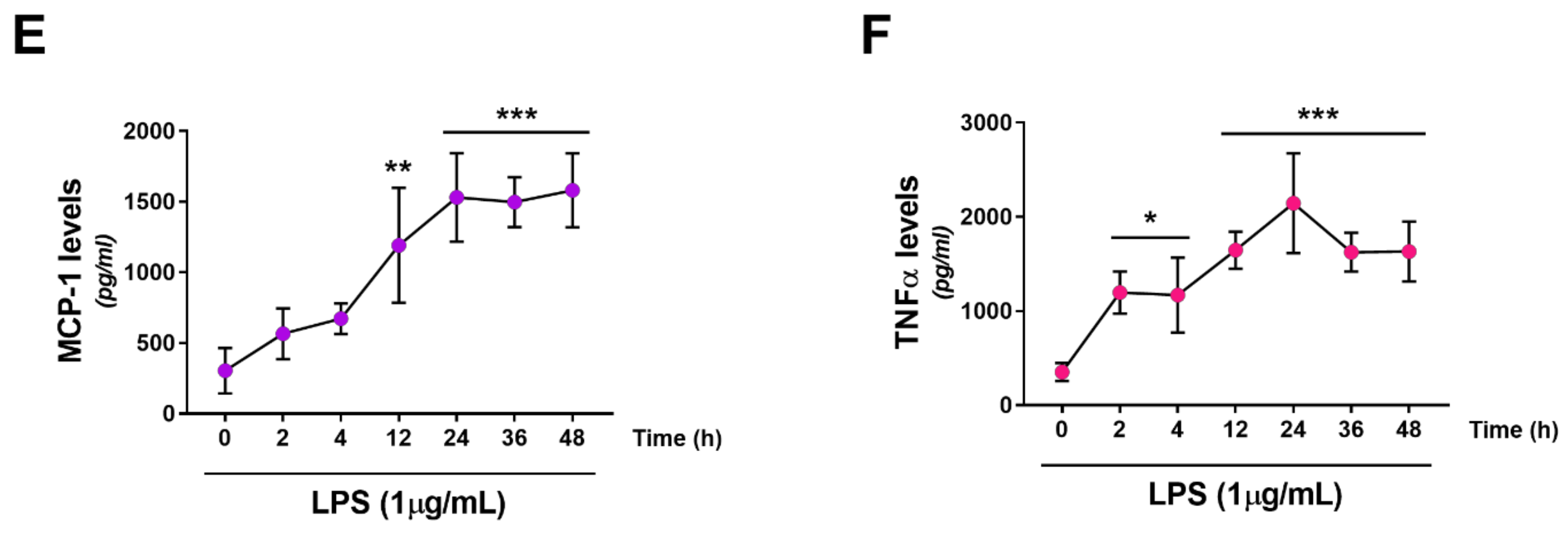
2.2. Effects of LPS Treatment on RT4 SCs Inflammatory Profile
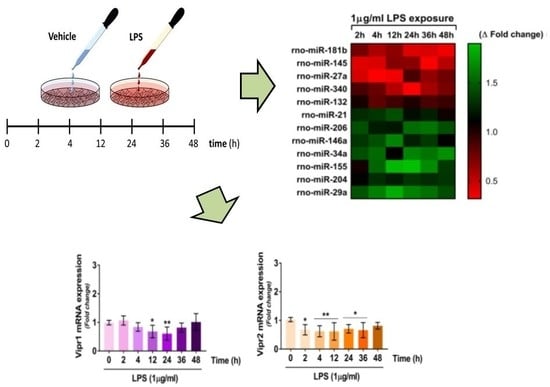
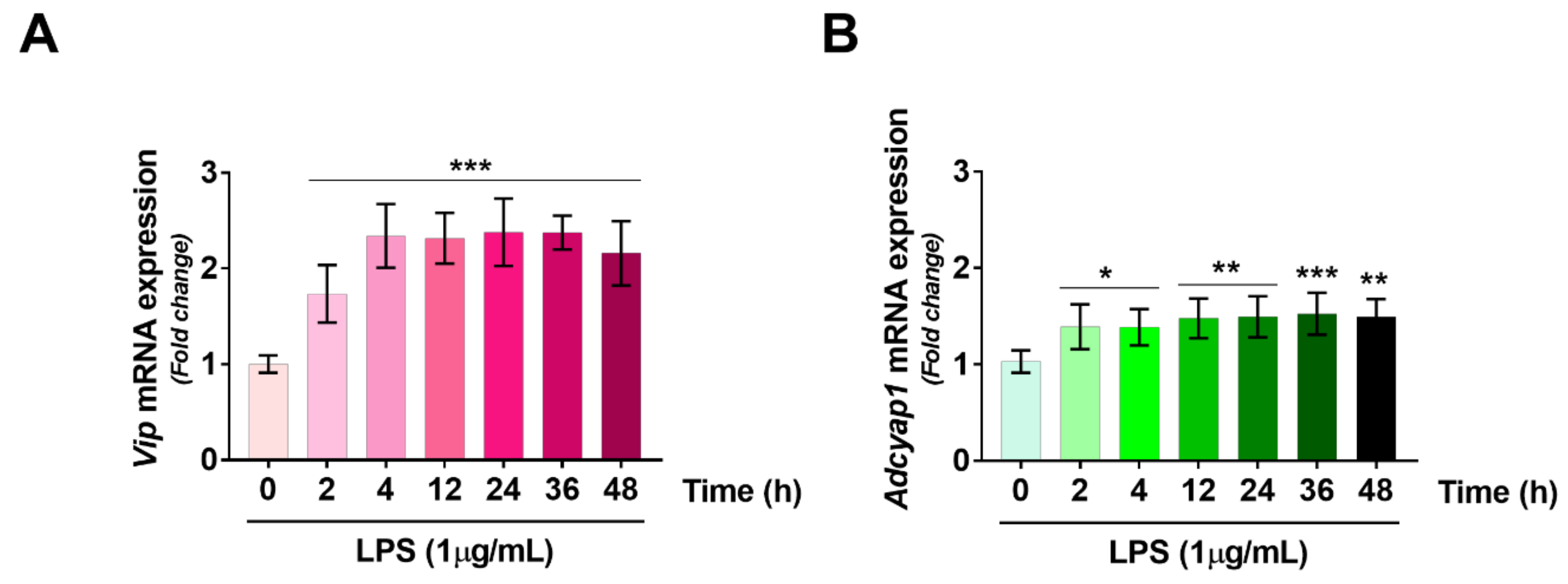
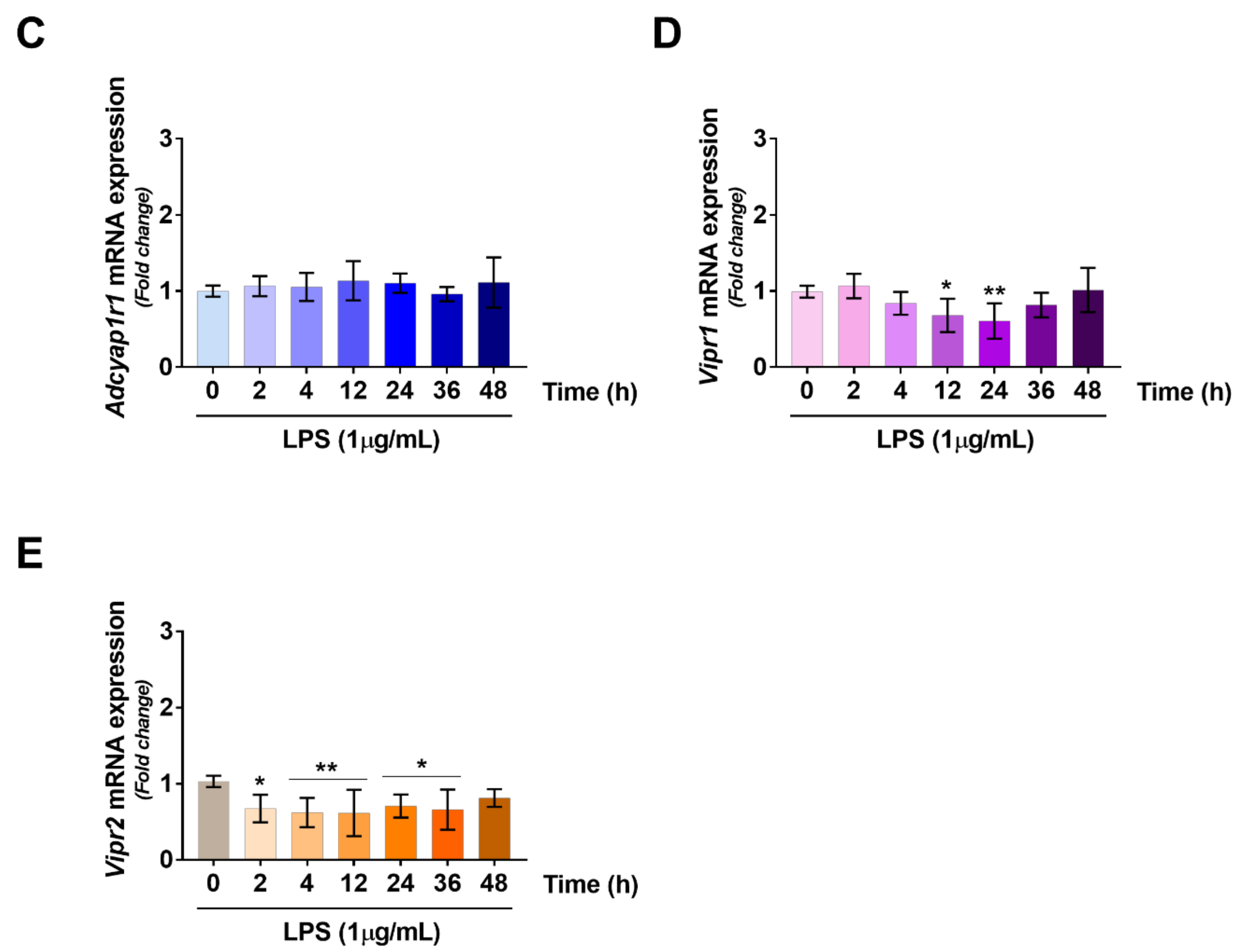
2.3. VIP/PACAP System Gene Expression Profile Following LPS Treatment
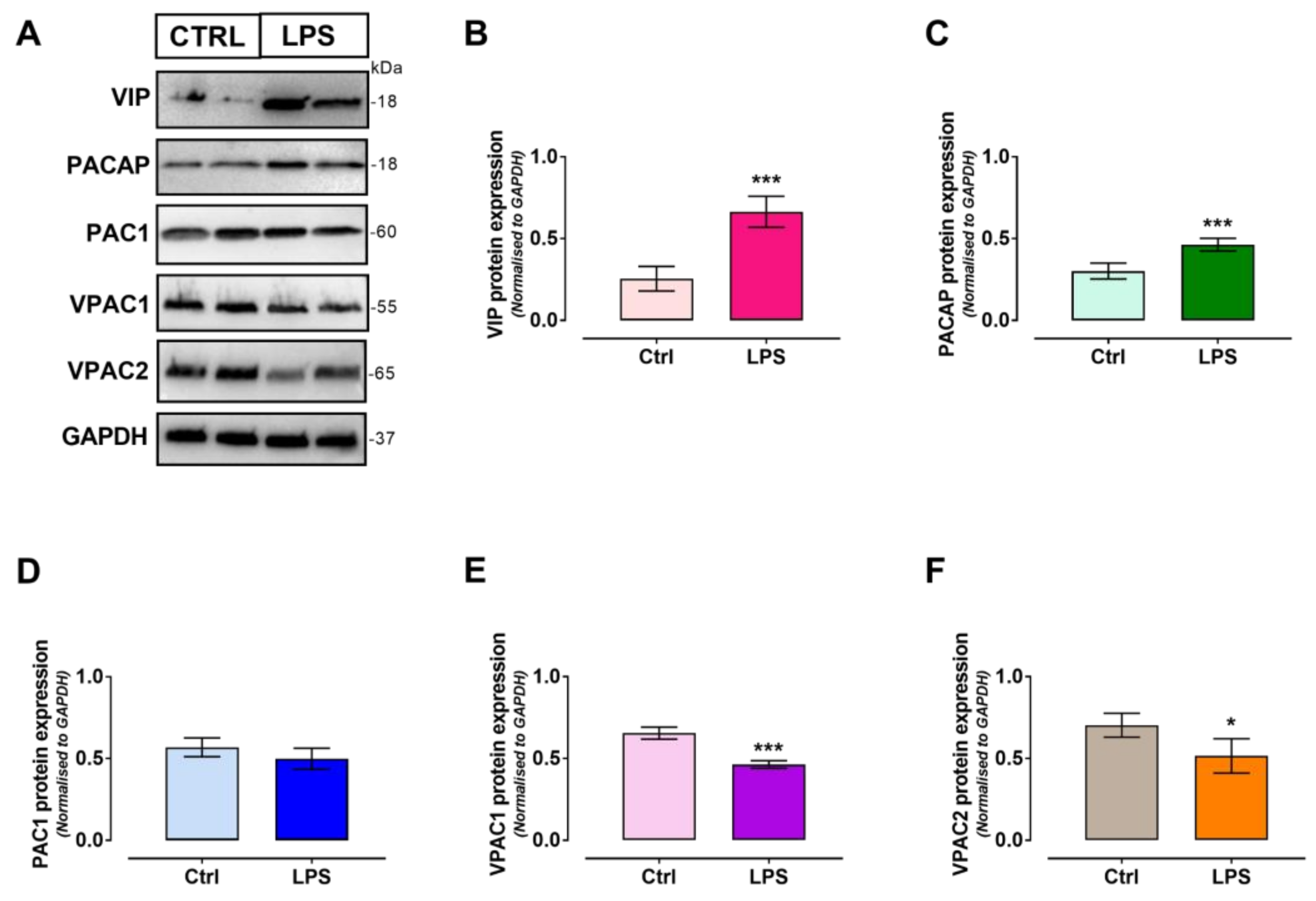
2.4. VIP/PACAP System Protein Expression Profile Following LPS Treatment
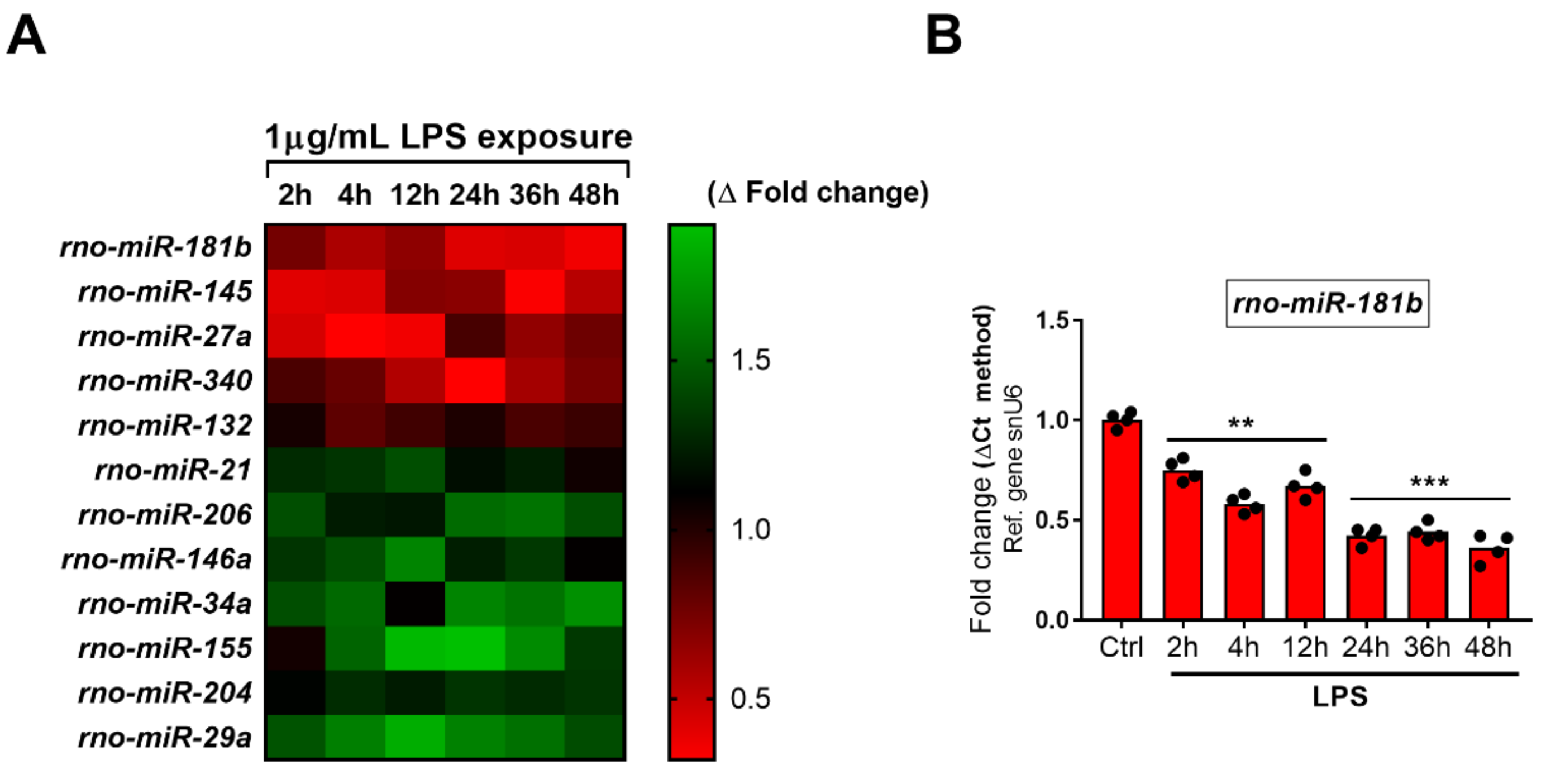
2.5. Dynamics of miRNAs Expression in RT4 SCs Exposed to LPS Treatment
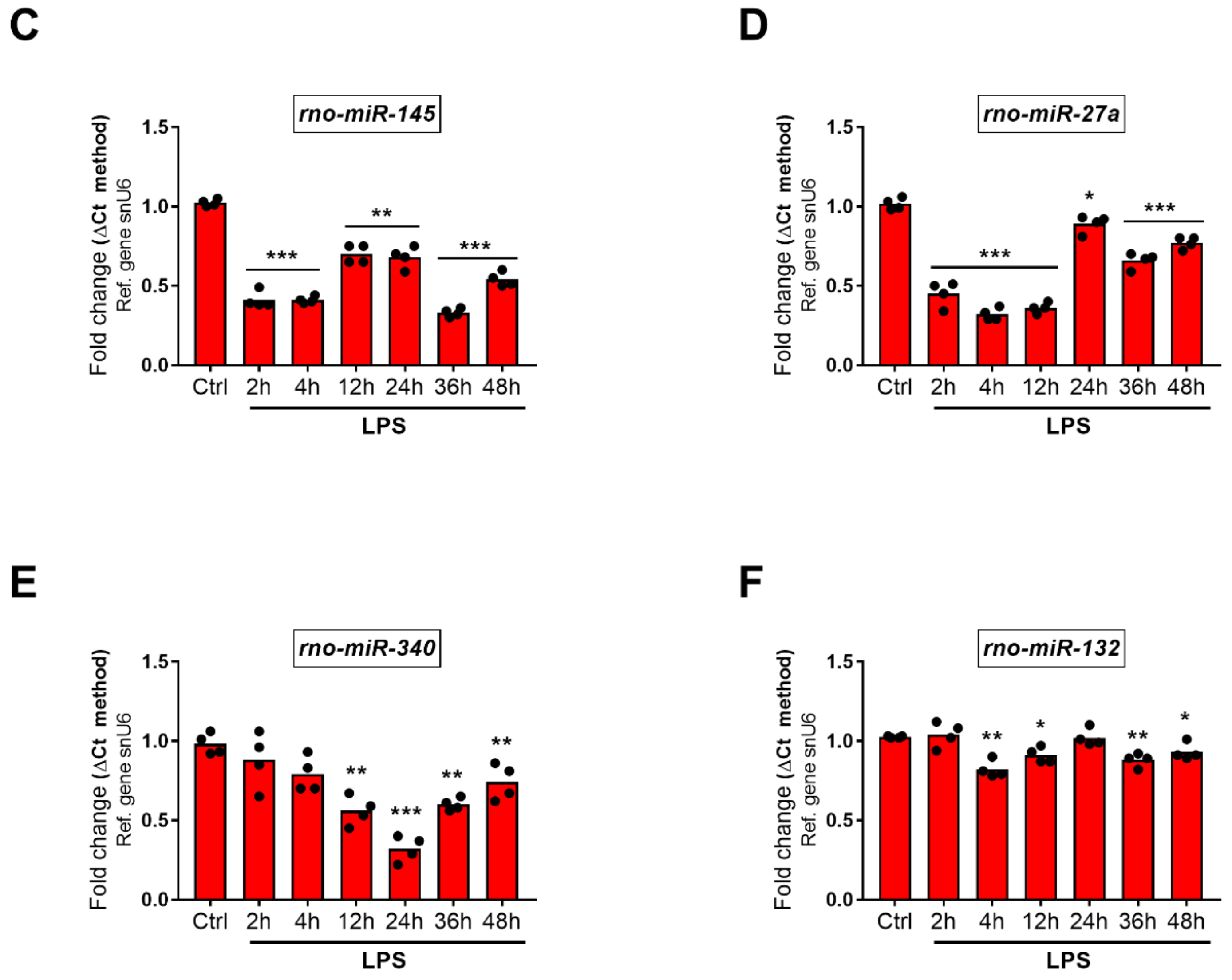
2.5.1. Downregulated miRNAs
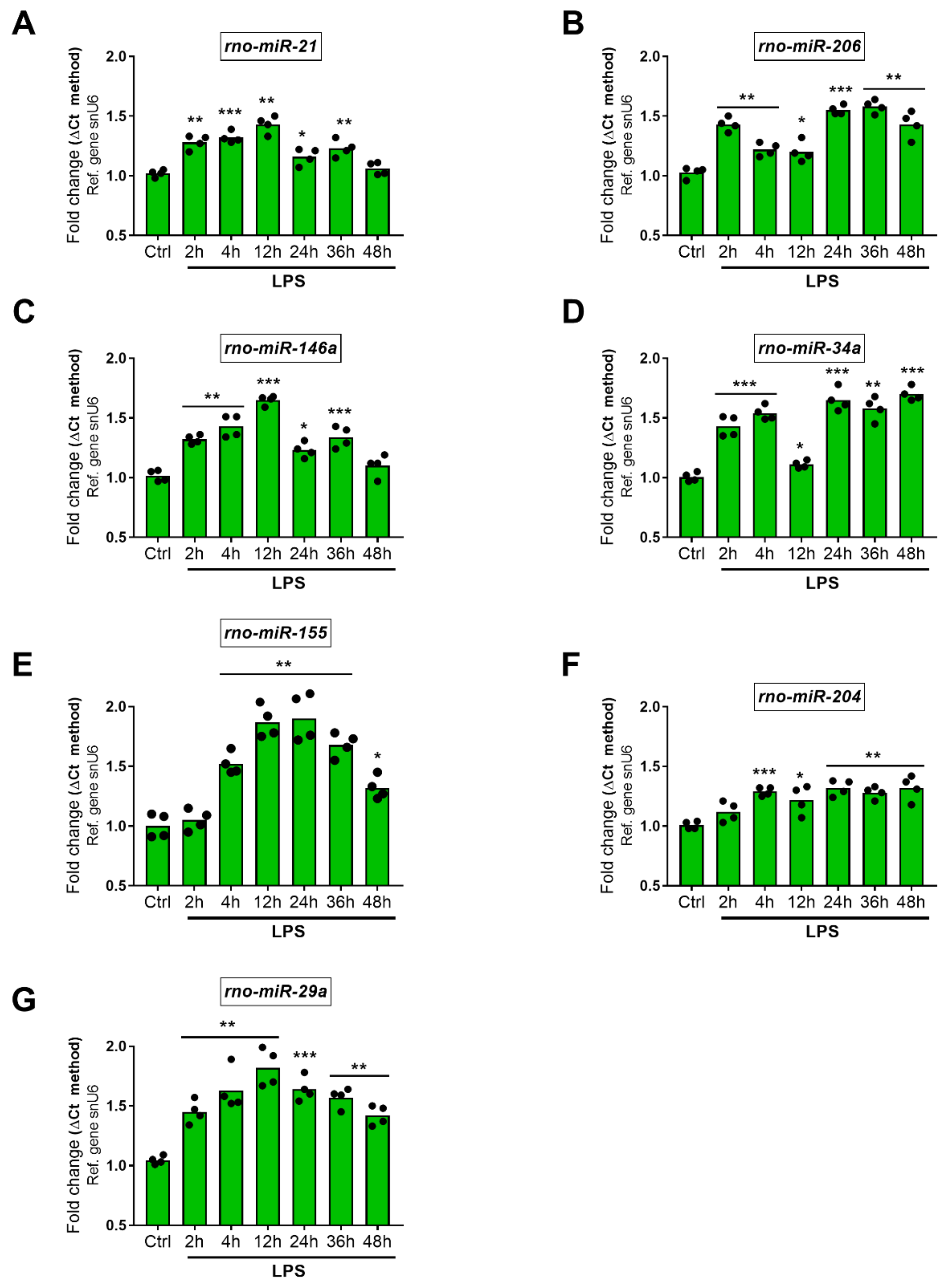
2.5.2. Upregulated miRNAs
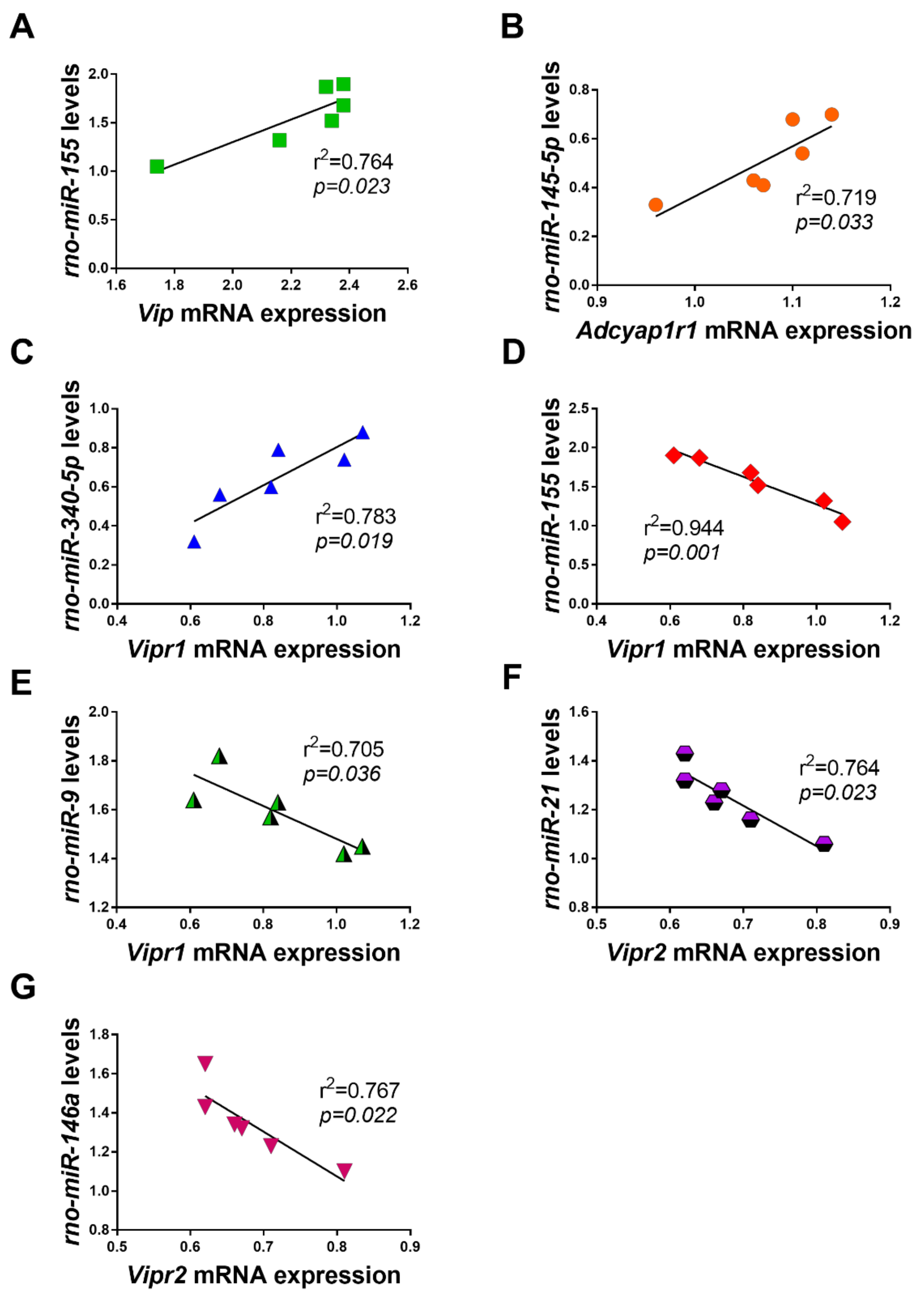
2.6. Correlations between Dysregulated miRNAs and VIP/PACAP System mRNAs in LPS Exposed RT4 SCs
3. Discussion
3.1. Dysregulated Expression of the VIP/PACAP System upon LPS Exposure
3.2. Dysregulated miRNAs and Their Potential Relationship with the VIP/PACAP System
4. Materials and Methods
4.1. RT4 Schwann Cell Line
4.2. Cell Viability Assay (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide Assay)
4.3. Hoechst 33258 Nuclear Staining
4.4. Multiplex Cytokine Assays
4.5. RNA Isolation, cDNA Synthesis and Gene Expression Analyses by Quantitative Real-Time PCR
4.6. SDS-Polyacrylamide Gel Electrophoresis and Western Blotting
4.7. MiRNA Profiling
4.8. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brosius Lutz, A.; Barres, B.A. Contrasting the glial response to axon injury in the central and peripheral nervous systems. Dev. Cell 2014, 28, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.D.; Talbot, W.S. Signals regulating myelination in peripheral nerves and the Schwann cell response to injury. Curr. Opin. Neurobiol. 2013, 23, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Scuderi, S.; D’Amico, A.G.; Drago, F.; D’Agata, V. PACAP and VIP increase the expression of myelin-related proteins in rat schwannoma cells: Involvement of PAC1/VPAC2 receptor-mediated activation of Pi3K/Akt signaling pathways. Exp. Cell Res. 2014, 322, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Waschek, J.A.; Marzagalli, R.; Cardile, V.; Drago, F. PACAP interacts with PAC1 receptors to induce tissue plasminogen activator (tPA) expression and activity in schwann cell-like cultures. PLoS ONE 2015, 10, e0117799. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Tiralongo, A.; Giunta, S.; Carnazza, M.L.; Rasi, G.; D’Agata, V. PACAP and VIP prevent apoptosis in schwannoma cells. Brain Res. 2008, 1241, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-V.; Waschek, J.A. Targeting VIP and PACAP receptor signalling: New therapeutic strategies in multiple sclerosis. Asn Neuro 2011, 3, e00065. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 Years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.G.; Scuderi, S.; Saccone, S.; Castorina, A.; Drago, F.; D’Agata, V. Antiproliferative effects of PACAP and VIP in serum-starved glioma cells. J. Mol. Neurosci. 2013, 51, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.P.; Byrne, E.; Camarillo Guerrero, L.F.; Cattin, A.-L.; Zakka, L.; Ashraf, A.; Burden, J.J.; Khadayate, S.; Lloyd, A.C.; Marguerat, S.; et al. The wound microenvironment reprograms schwann cells to invasive mesenchymal-like cells to drive peripheral nerve regeneration. Neuron 2017, 96, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Wu, L.; Belasco, J.G. Let me count the ways: Mechanisms of gene regulation by miRNAs and siRNAs. Mol. Cell 2008, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Vimal, D.; Sharma, D.; Rai, V.; Gupta, S.C.; Chowdhuri, D.K. Role of miRNAs in development and disease: Lessons learnt from small organisms. Life Sci. 2017, 185, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005, 6, 671–682. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, M.; Michalovich, D.; Paterson, M.; Droggiti, A.; Woodhoo, A.; Mirsky, R.; Jessen, K.R. Gene profiling and bioinformatic analysis of Schwann cell embryonic development and myelination. Glia 2006, 53, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Ydens, E.; Lornet, G.; Smits, V.; Goethals, S.; Timmerman, V.; Janssens, S. The neuroinflammatory role of Schwann cells in disease. Neurobiol. Dis. 2013, 55, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhou, S.; Wang, Y.; Qian, T.; Ding, G.; Ding, F.; Gu, X. miR-221 and miR-222 promote Schwann cell proliferation and migration by targeting LASS2 after sciatic nerve injury. J. Cell Sci. 2012, 125, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Li, X.; Li, Y.; Che, J.; Li, X.; Zhao, X.; Chen, Y.; Zheng, X.; Yuan, W. Biodegradable and biocompatible cationic polymer delivering microRNA-221/222 promotes nerve regeneration after sciatic nerve crush. Int. J. Nanomed. 2017, 12, 4195–4208. [Google Scholar] [CrossRef] [PubMed]
- Verrier, J.D.; Lau, P.; Hudson, L.; Murashov, A.K.; Renne, R.; Notterpek, L. Peripheral myelin protein 22 is regulated post-transcriptionally by miRNA-29a. Glia 2009, 57, 1265–1279. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, T.; Yuan, H.; Han, X.; Shui, S.; Guo, D.; Yan, L. MicroRNA-29a inhibits proliferation and motility of Schwannoma cells by targeting CDK6. J. Cell. Biochem. 2018, 119, 2617–2626. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zhang, K.; Liu, X.; Yang, T.; Wang, B.; Fu, L. Mir-21 promotes the differentiation of hair follicle-derived neural crest stem cells into schwann cells. Neural Regen. Res. 2014, 9, 828–836. [Google Scholar] [PubMed]
- Zhou, S.; Gao, R.; Hu, W.; Qian, T.; Wang, N.; Ding, G.; Ding, F.; Yu, B.; Gu, X. Mir-9 inhibits schwann cell migration by targeting cthrc1 following sciatic nerve injury. J. Cell Sci. 2014, 127, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Sun, C.; Hu, W.; Zhao, J.; Li, G.; Zhang, L.; Liu, M.; Liu, Y.; Ding, F.; et al. Chitosan degradation products promote nerve regeneration by stimulating schwann cell proliferation via mir-27a/foxo1 axis. Mol. Neurobiol. 2016, 53, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Viader, A.; Chang, L.W.; Fahrner, T.; Nagarajan, R.; Milbrandt, J. Micrornas modulate schwann cell response to nerve injury by reinforcing transcriptional silencing of dedifferentiation-related genes. J. Neurosci. 2011, 31, 17358–17369. [Google Scholar] [CrossRef] [PubMed]
- Truettner, J.S.; Motti, D.; Dietrich, W.D. Microrna overexpression increases cortical neuronal vulnerability to injury. Brain Res. 2013, 1533, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Shi, X.; Zhang, Z.; Zhou, S.; Qian, T.; Wang, Y.; Ding, F.; Gu, X.; Yu, B. Hypoxia-induced upregulation of mir-132 promotes schwann cell migration after sciatic nerve injury by targeting prkag3. Mol. Neurobiol. 2016, 53, 5129–5139. [Google Scholar] [CrossRef] [PubMed]
- Verrier, J.D.; Semple-Rowland, S.; Madorsky, I.; Papin, J.E.; Notterpek, L. Reduction of dicer impairs schwann cell differentiation and myelination. J. Neurosci. Res. 2010, 88, 2558–2568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gong, X.; Qiu, J.; Zhang, Y.; Gong, F. Microrna-210 contributes to peripheral nerve regeneration through promoting the proliferation and migration of schwann cells. Exp. Ther. Med. 2017, 14, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Gokey, N.G.; Srinivasan, R.; Lopez-Anido, C.; Krueger, C.; Svaren, J. Developmental regulation of microrna expression in schwann cells. Mol. Cell. Biol. 2012, 32, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Garo, L.P.; Murugaiyan, G. The use of mirna antagonists in the alleviation of inflammatory disorders. Methods Mol. Biol. 2016, 1390, 413–425. [Google Scholar] [PubMed]
- Hutchison, E.R.; Kawamoto, E.M.; Taub, D.D.; Lal, A.; Abdelmohsen, K.; Zhang, Y.; Wood, W.H., 3rd.; Lehrmann, E.; Camandola, S.; Becker, K.G.; et al. Evidence for mir-181 involvement in neuroinflammatory responses of astrocytes. Glia 2013, 61, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Qian, T.; Wang, Y.; Zhou, S.; Ding, G.; Ding, F.; Gu, X. Mir-182 inhibits schwann cell proliferation and migration by targeting fgf9 and ntm, respectively at an early stage following sciatic nerve injury. Nucleic Acids Res. 2012, 40, 10356–10365. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wang, L.; Sun, J.; Nie, K.; Jian, H.; Gao, L.; Liao, X.; Zhang, H.; Huang, J.; Gan, S. Mir-204 promotes apoptosis in oxidative stress-induced rat schwann cells by suppressing neuritin expression. FEBS Lett. 2014, 588, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Torres-Martin, M.; Lassaletta, L.; de Campos, J.M.; Isla, A.; Gavilan, J.; Pinto, G.R.; Burbano, R.R.; Latif, F.; Melendez, B.; Castresana, J.S.; et al. Global profiling in vestibular schwannomas shows critical deregulation of micrornas and upregulation in those included in chromosomal region 14q32. PLoS ONE 2013, 8, e65868. [Google Scholar] [CrossRef] [PubMed]
- Paschou, M.; Doxakis, E. Neurofibromin 1 is a mirna target in neurons. PLoS ONE 2012, 7, e46773. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Dai, C.; Feng, Z.; Zhang, L.; Zhang, Z. Mir-137 inhibited inflammatory response and apoptosis after spinal cord injury via targeting of mk2. J. Cell. Biochem. 2018, 119, 3280–3292. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, R.; Yuan, Y.; Yi, S.; Chen, Q.; Gong, L.; Liu, J.; Ding, F.; Cao, Z.; Gu, X. Mir-340 regulates fibrinolysis and axon regrowth following sciatic nerve injury. Mol. Neurobiol. 2017, 54, 4379–4389. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N. Microrna biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Giunta, S.; Scuderi, S.; D’Agata, V. Involvement of pacap/adnp signaling in the resistance to cell death in malignant peripheral nerve sheath tumor (mpnst) cells. J. Mol. Neurosci. 2012, 48, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Lopez, J.P.; Laufer, S.A.; Miao, E.A. Il-1beta, il-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur. J. Immunol. 2016, 46, 2761–2766. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.L.; Berk, E.; Edwards, R.P.; Kalinski, P. Il-18-primed helper nk cells collaborate with dendritic cells to promote recruitment of effector cd8+ t cells to the tumor microenvironment. Cancer Res. 2013, 73, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Uceyler, N.; Tscharke, A.; Sommer, C. Early cytokine gene expression in mouse cns after peripheral nerve lesion. Neurosci. Lett. 2008, 436, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Brenneman, D.E. Neuroprotection: A comparative view of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Peptides 2007, 28, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Waschek, J.A. Vip and pacap: Neuropeptide modulators of cns inflammation, injury, and repair. Br. J. Pharmacol. 2013, 169, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J.; Fahrenkrug, J.; Gozes, I.; Laburthe, M.; May, V.; Pisegna, J.R.; Vaudry, D.; Vaudry, H.; Waschek, J.A.; Said, S.I. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: Iuphar review 1. Br. J. Pharmacol. 2012, 166, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Waschek, J.A. Multiple actions of pituitary adenylyl cyclase activating peptide in nervous system development and regeneration. Dev. Neurosci. 2002, 24, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Canas, I.; Rodriguez-Henche, N.; Bolanos, O.; Carmena, M.J.; Prieto, J.C.; Juarranz, M.G. Vip and pacap are autocrine factors that protect the androgen-independent prostate cancer cell line pc-3 from apoptosis induced by serum withdrawal. Br. J. Pharmacol. 2003, 139, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, M.; Furuta, A.; Aoki, S.; Kudo, Y.; Miyakawa, H.; Wada, K. Pacap/pac1 autocrine system promotes proliferation and astrogenesis in neural progenitor cells. Glia 2007, 55, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. Negative regulation of myelination: Relevance for development, injury, and demyelinating disease. Glia 2008, 56, 1552–1565. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Shin, Y.K.; Park, H.T. Mitogen activated protein kinase family proteins and c-jun signaling in injury-induced schwann cell plasticity. Exp. Neurobiol. 2014, 23, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Nicolino, S.; Tos, P.; Battiston, B.; Giacobini-Robecchi, M.G.; Perroteau, I.; Geuna, S. Schwann cell behavior after nerve repair by means of tissue-engineered muscle-vein combined guides. J. Comp. Neurol. 2005, 489, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Oya, T.; Zhao, Y.L.; Takagawa, K.; Kawaguchi, M.; Shirakawa, K.; Yamauchi, T.; Sasahara, M. Platelet-derived growth factor-b expression induced after rat peripheral nerve injuries. Glia 2002, 38, 303–312. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. Microrna-155 promotes autoimmune inflammation by enhancing inflammatory t cell development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Billeter, A.T.; Hellmann, J.; Roberts, H.; Druen, D.; Gardner, S.A.; Sarojini, H.; Galandiuk, S.; Chien, S.; Bhatnagar, A.; Spite, M.; et al. Microrna-155 potentiates the inflammatory response in hypothermia by suppressing il-10 production. FASEB J. 2014, 28, 5322–5336. [Google Scholar] [CrossRef] [PubMed]
- Dugas, J.C.; Notterpek, L. Micrornas in oligodendrocyte and schwann cell differentiation. Dev. Neurosci. 2011, 33, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Giunta, S.; Mazzone, V.; Cardile, V.; D’Agata, V. Effects of pacap and vip on hyperglycemia-induced proliferation in murine microvascular endothelial cells. Peptides 2010, 31, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Leggio, G.M.; Giunta, S.; Magro, G.; Scapagnini, G.; Drago, F.; D’Agata, V. Neurofibromin and amyloid precursor protein expression in dopamine d3 receptor knock-out mice brains. Neurochem. Res. 2011, 36, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time pcr data by the comparative c(t) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, S.; D’Amico, A.G.; Castorina, A.; Federico, C.; Marrazzo, G.; Drago, F.; Bucolo, C.; D’Agata, V. Davunetide (nap) protects the retina against early diabetic injury by reducing apoptotic death. J. Mol. Neurosci. 2014, 54, 395–404. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Ref. seq.) | Primers | Location of Primers | Tm (°C) | Length (bp) |
|---|---|---|---|---|
| Rattus norvegicus adenylate cyclase activating polypeptide 1 (Adcyap1) (NM_016989.2) | 5′-GAGGCTTACGATCAGGACGG-3′ | 414 | 59.97 | 121 |
| 3′-TCCTGTCGGCTGGGTAGTAA-5′ | 534 | 59.96 | ||
| Rattus norvegicus vasoactive intestinal peptide (Vip) (NM_053991.1) | 5′-GTAGCATCTCGGAAGACCCC-3′ | 481 | 59.61 | 83 |
| 3′-TTGCTTTCTAAGGCGGGTGT-5′ | 563 | 59.89 | ||
| Rattus norvegicus ADCYAP receptor type 1 (Adcyap1r1) (NM_001270579.1) | 5′-GACCAGCATTCACCCCCTTT-3′ | 1442 | 60.25 | 114 |
| 3′-CAGCCGTAGAGTAATGGTGGAT-5′ | 1555 | 59.63 | ||
| Rattus norvegicus vasoactive intestinal peptide receptor 1 (Vipr1) (NM_012685.2) | 5′-AAGCTGCACTGTACCCGAAA-3′ | 597 | 59.89 | 103 |
| 3′-CGCTGTTGAAGAGGGCCATA-5′ | 699 | 60.11 | ||
| Rattus norvegicus vasoactive intestinal peptide receptor 2 (Vipr2) (NM_017238.1) | 5′-TGACCTGCTACTGCTGGTTG-3′ | 135 | 59.96 | 138 |
| 3′-CGCTGCAAGCTCTGTGATTC-5′ | 272 | 59.9 | ||
| Rattus norvegicus ribosomal protein S18 (Rps18) (NM_213557.1) | 5′-AGCGGCTGAAGAAAATCCGA-3′ | 380 | 60.04 | 115 |
| 3′-TTGGACACACCCACAGTACG-5′ | 494 | 59.89 |
| miRNA | Putative Role in SCs | Identified Targets | Reference(s) |
|---|---|---|---|
| miR-221 | Cell migration, myelin genes | LAAS2 | [18,19] |
| miR-222 | Cell migration, myelin genes | LAAS2 | [18,19] |
| miR-29a | Myelin genes, motility | PMP-22, CDK6 | [20,21] |
| miR-21 | Cell differentiation | SOX-2 | [22] |
| miR-9 | Cell migration | Cthrc1 | [23] |
| miR-27a | Cell proliferation | FOXO1 | [24] |
| miR-34a | De-differentiation genes, apoptosis | Notch1, Ccnd1, Bcl2, XLAP | [25,26] |
| miR-132 | Cell migration | PRKAG3 | [27] |
| miR-145 | De-differentiation genes | Egr2, c-Jun, MPZ | [25,28] |
| miR-210 | Cell proliferation & migration | GAP-43, MAG, MBP | [29] |
| miR-146a | Schwann cell development | SOX-10 | [30] |
| miR-155 | Inflammatory response | Toll-like receptors | [31] |
| miR-181b | Inflammation (in astrocytes) | MeCP2, XLAP | [32] |
| miR-182 | Cell proliferation and migration | FGF9, NTM | [33] |
| miR-204 | Apoptotic process | Neuritin | [34] |
| miR-206 | Oncogenic transformation | NF2, ERBB2, NRG1 | [35] |
| miR-137 | Tumor suppressor function | NF1, MK2 | [36,37] |
| miR-340 | Production of proteolytic enzymes | tPA | [38] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musumeci, G.; Leggio, G.M.; Marzagalli, R.; Al-Badri, G.; Drago, F.; Castorina, A. Identification of Dysregulated microRNA Networks in Schwann Cell-Like Cultures Exposed to Immune Challenge: Potential Crosstalk with the Protective VIP/PACAP Neuropeptide System. Int. J. Mol. Sci. 2018, 19, 981. https://doi.org/10.3390/ijms19040981
Musumeci G, Leggio GM, Marzagalli R, Al-Badri G, Drago F, Castorina A. Identification of Dysregulated microRNA Networks in Schwann Cell-Like Cultures Exposed to Immune Challenge: Potential Crosstalk with the Protective VIP/PACAP Neuropeptide System. International Journal of Molecular Sciences. 2018; 19(4):981. https://doi.org/10.3390/ijms19040981
Chicago/Turabian StyleMusumeci, Giuseppe, Gian Marco Leggio, Rubina Marzagalli, Ghaith Al-Badri, Filippo Drago, and Alessandro Castorina. 2018. "Identification of Dysregulated microRNA Networks in Schwann Cell-Like Cultures Exposed to Immune Challenge: Potential Crosstalk with the Protective VIP/PACAP Neuropeptide System" International Journal of Molecular Sciences 19, no. 4: 981. https://doi.org/10.3390/ijms19040981
APA StyleMusumeci, G., Leggio, G. M., Marzagalli, R., Al-Badri, G., Drago, F., & Castorina, A. (2018). Identification of Dysregulated microRNA Networks in Schwann Cell-Like Cultures Exposed to Immune Challenge: Potential Crosstalk with the Protective VIP/PACAP Neuropeptide System. International Journal of Molecular Sciences, 19(4), 981. https://doi.org/10.3390/ijms19040981